I am new regarding the KEGG pathway enrichment analysis. I have an in-house assembled genome and used it for RNAseq analysis. I did KEGG annotation using GhostKOALA and then used the KO annotation for GSEA analysis using ClusterProfiler (version 4.4.4). It ran smoothly but the enrichment result was a list of KO entries. I guess it makes sense since I used this KO annotation as the TERM2GENE input (KO used as the term). However, what I am interested in is enriched KEGG pathways and their interactions. Any suggestions on how to get that? Do I need to use KEGG mapped pathway code as the term used in TERM2GENE? I used KEGG mapper to reconstruct the pathway based on the KO annotation, but haven't found a way on transforming the on-line result from https://www.genome.jp/kegg/mapper/reconstruct.html into the TERM2GENE format. Any suggestions would be greatly appreciated.
I don't fully understand your question. I am confused by what you mean with KO entries in "... but the enrichment result was a list of KO entries."
So, in essence, you have a list of enriched pathways that are identified by a ko-id (ko in lower case), such as ko01200? And you now would like to know it is the KEGG gene set Carbon metabolism? https://www.genome.jp/dbget-bin/www_bget?ko01200
If so, you can make use of the function download_KEGG. It will return 2 data.frames, that you can use for the arguments TERM2GENE and TERM2NAME when running the universal function GSEA (which you likely did, but then only with 'your' TERM2GENE as input).
If not, please show some reproducible code that illustrates what you try to achieve.
Thank you so much for your reply! My apologies that I didn't make it clear. GhostKOALA annotation returns a list of KEGG Orthology (KO) entries for the genes such as K00850 (https://www.kegg.jp/dbget-bin/www_bget?K00850 ). For GSEA analysis, I have occasions such as below: Multiple unique genes from the genome with different fold changes all have the same KO entry.
There are also warnings from GSEA run "2: In preparePathwaysAndStats(pathways, stats, minSize, maxSize, gseaParam, :
There are duplicate gene names, fgsea may produce unexpected results."
Therefore, I opted to use the unique gene IDs from the assembled genome as the gene (term2gene), and the KO entries above as the term.
As a result, my enriched result would be the terms I have in the TERM2GENE list. Your answer cleared my first question that I need to use ko terms in TERM2GENE (Thanks). So, I guess my question now would be how to transform the KO entries I have into the ko terms, so I can replace the KO entries from the TERM2GENE list to ko terms you mentioned, but still maintain/use my unique gene ID in the GSEA input list. I am not sure if this makes sense.
So if I understand correctly the same gene (KOxxxxx) is present multiple times in your (ranked) input, and for each entry the logFC is different.
This is indeed the reason that fgsea warns you with: There are duplicate gene names, fgsea may produce unexpected results.
I don't have any experience with GhostKOALA, so I cannot recommend on the use of that tool, but I would check why multiple genes map to the same gene/KO. Would it not best to keep the gene/KO that has the lowest BLAST score (= most specific)? Or take the mean, or median of all logFC for a gene? Or ...?
BTW: this actually is a classic question in the context of microarray analysis... See e.g. here or here.
Thank you very much for your suggestions and the link to other related posts. I looked into these genes and they belong to the same transcription factor family, so I think it would be reasonable to have differences on their expression levels. Not sure what the best practices in this situation but I am tempted to ignore them in the GSEA analysis.
I have a similar issue, I have transcriptome data of an inhouse sequenced bacterial genome. I made the database for that bacteria using makeOrgPackageFromNCBI command.
How can I use this database for KEGG AND Go analysis (ORA or GSEA). All commands use either KEGG organism database for such an analysis. I tried with that command but it saved "Gene ids did not match" as output.
If I want to use the above method, by mapping Ko ids as Term2gene, how can I extract term2name database. Can I take from the closet species available in KEGG database.
If KEGG database of a closet species (with term2gene and term2name) for analysis and I have identified the KO ids of my filtered DEGs using eggnog, but for some gene such as sRNAs or hypothetical protein, I do not have the Ko ids. I will miss those gene ids from my analysis?
I asked you in a previous thread to not add comments to old threads and instead start a new one. Please follow that advice. It's fine if you want to reference an old thread, but please stop resurrecting them like this.


I don't fully understand your question. I am confused by what you mean with KO entries in "... but the enrichment result was a list of KO entries."
So, in essence, you have a list of enriched pathways that are identified by a ko-id (ko in lower case), such as
ko01200? And you now would like to know it is the KEGG gene setCarbon metabolism? https://www.genome.jp/dbget-bin/www_bget?ko01200If so, you can make use of the function
download_KEGG. It will return 2data.frames, that you can use for the argumentsTERM2GENEandTERM2NAMEwhen running the universal functionGSEA(which you likely did, but then only with 'your'TERM2GENEas input).If not, please show some reproducible code that illustrates what you try to achieve.
Thank you so much for your reply! My apologies that I didn't make it clear. GhostKOALA annotation returns a list of KEGG Orthology (KO) entries for the genes such as K00850 (https://www.kegg.jp/dbget-bin/www_bget?K00850 ). For GSEA analysis, I have occasions such as below: Multiple unique genes from the genome with different fold changes all have the same KO entry.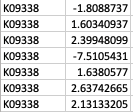 There are also warnings from GSEA run "2: In preparePathwaysAndStats(pathways, stats, minSize, maxSize, gseaParam, :
There are duplicate gene names, fgsea may produce unexpected results."
Therefore, I opted to use the unique gene IDs from the assembled genome as the gene (term2gene), and the KO entries above as the term.
There are also warnings from GSEA run "2: In preparePathwaysAndStats(pathways, stats, minSize, maxSize, gseaParam, :
There are duplicate gene names, fgsea may produce unexpected results."
Therefore, I opted to use the unique gene IDs from the assembled genome as the gene (term2gene), and the KO entries above as the term.
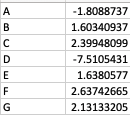
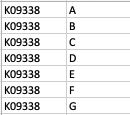 As a result, my enriched result would be the terms I have in the TERM2GENE list. Your answer cleared my first question that I need to use ko terms in TERM2GENE (Thanks). So, I guess my question now would be how to transform the KO entries I have into the ko terms, so I can replace the KO entries from the TERM2GENE list to ko terms you mentioned, but still maintain/use my unique gene ID in the GSEA input list. I am not sure if this makes sense.
As a result, my enriched result would be the terms I have in the TERM2GENE list. Your answer cleared my first question that I need to use ko terms in TERM2GENE (Thanks). So, I guess my question now would be how to transform the KO entries I have into the ko terms, so I can replace the KO entries from the TERM2GENE list to ko terms you mentioned, but still maintain/use my unique gene ID in the GSEA input list. I am not sure if this makes sense.
So if I understand correctly the same gene (KOxxxxx) is present multiple times in your (ranked) input, and for each entry the logFC is different.
This is indeed the reason that
fgseawarns you with:There are duplicate gene names, fgsea may produce unexpected results.I don't have any experience with GhostKOALA, so I cannot recommend on the use of that tool, but I would check why multiple genes map to the same gene/KO. Would it not best to keep the gene/KO that has the lowest BLAST score (= most specific)? Or take the mean, or median of all logFC for a gene? Or ...?
BTW: this actually is a classic question in the context of microarray analysis... See e.g. here or here.
Thank you very much for your suggestions and the link to other related posts. I looked into these genes and they belong to the same transcription factor family, so I think it would be reasonable to have differences on their expression levels. Not sure what the best practices in this situation but I am tempted to ignore them in the GSEA analysis.