
Hello!
This is my first attempt to do DESeq2 analysis. I followed the vignettes and tutorials I could find online as best, but got unexpectedly low number of DE genes. In the experiment, there are 2 subjects (X90 and X92) and 2 paired samples from each (Tom=control and Venus=treated). The counts were obtained using salmon, and for the DESeq2 analysis (DESeq2_1.36.0) I have used the salmon.merged.gene_counts_length_scaled.tsv, as recommended.
salmon_table<-read.table("results/star_salmon/salmon.merged.gene_counts_length_scaled.tsv",
sep="\t",header=TRUE,row.names="gene_id")
cts <- salmon_table[,c(2:5)] # sub-setting to exclude a subject with problematic sequencing quality
cts <- as.matrix(round(cts))
coldata<-read.csv("metadata.csv",header=TRUE,row.names=1)
coldata<-coldata[1:4,] # sub-setting to exclude a subject with problematic sequencing quality
dds<- DESeqDataSetFromMatrix(countData = cts,
colData = coldata,
design= ~ mouse+genotype)
dds <- dds[ rowSums(counts(dds)) > 1, ]
vsd<-vst(dds, blind=TRUE)
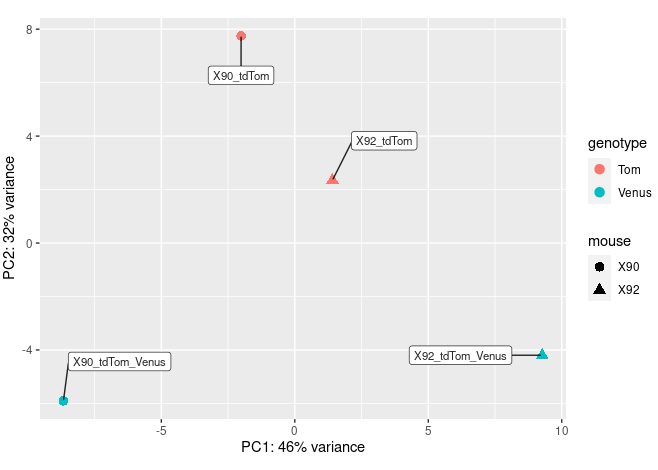
pcaData <- plotPCA(vsd, intgroup=c("mouse","genotype"), returnData=TRUE)
percentVar <- round(100 * attr(pcaData, "percentVar"))
ggplot(pcaData, aes(PC1, PC2, color=genotype, shape=mouse)) +
geom_point(size=3) +
xlab(paste0("PC1: ",percentVar[1],"% variance")) +
ylab(paste0("PC2: ",percentVar[2],"% variance")) +
coord_fixed() +
geom_label_repel(aes(label = name), colour = I(alpha("black", 0.85)),
size = 3,box.padding = 1.5,fill="white",max.overlaps = 30 );
dds<-DESeq(dds)
res_genotype_Venus_vs_Tom_noshrink<- results(dds, contrast=c("genotype","Venus","Tom"), independentFiltering=TRUE)
summary(res_genotype_Venus_vs_Tom_noshrink)
out of 30674 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 12, 0.039%
LFC < 0 (down) : 57, 0.19%
outliers [1] : 0, 0%
low counts [2] : 16057, 52%
(mean count < 20)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?results
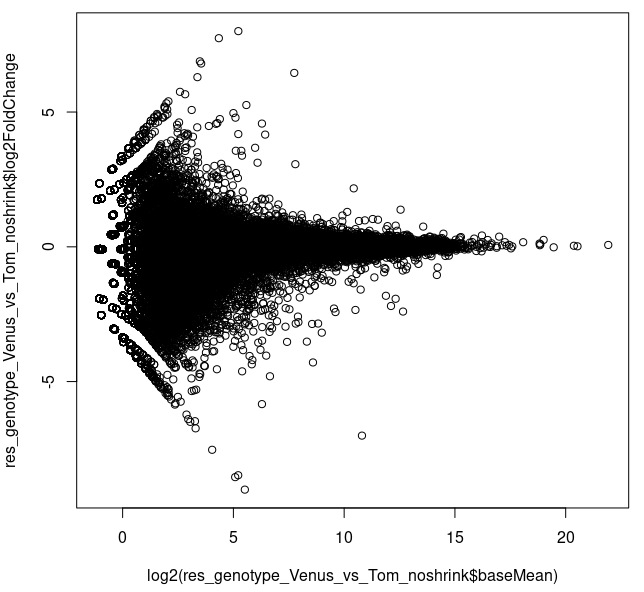
About half (52%) of the genes were filtered due to low counts (<20). Looking at the MA plot, it appears that there are some genes high-ish log2FC exhibiting such average counts.
plot(log2(res_genotype_Venus_vs_Tom_noshrink$baseMean),
res_genotype_Venus_vs_Tom_noshrink$log2FoldChange)
I had to plot it as above, since the default plotting command plotMA() produced
plotMA(res_genotype_Venus_vs_Tom_noshrink)
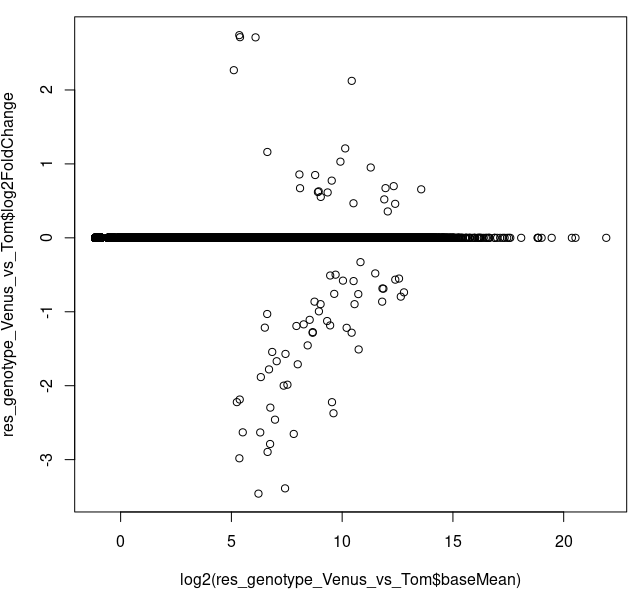
I also performed shrinking and it looks like the genes with < 2^^5 = 32 counts have shrunk log2FC set to ~0 (which, from what I read online can be due to either "low counts" or "high dispersion")
res_genotype_Venus_vs_Tom <- lfcShrink(dds, coef="genotype_Venus_vs_Tom",
type="apeglm",res=res_genotype_Venus_vs_Tom_noshrink)
plot(log2(res_genotype_Venus_vs_Tom$baseMean),
res_genotype_Venus_vs_Tom$log2FoldChange)
My questions are: am I doing something wrong? Am I missing some DE genes from the mean count < 20 region? And how DESeq2 arrived at the cut-off value of 20?
Thank you in advance for any insight/suggestions!
sessionInfo( )
R version 4.2.0 (2022-04-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.5 (Nitrogen)
Matrix products: default
BLAS: /exports/eddie3_apps_local/apps/community/roslin/R/4.2.0/lib64/R/lib/libRblas.so
LAPACK: /exports/eddie3_apps_local/apps/community/roslin/R/4.2.0/lib64/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_GB.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_GB.UTF-8 LC_COLLATE=en_GB.UTF-8
[5] LC_MONETARY=en_GB.UTF-8 LC_MESSAGES=en_GB.UTF-8
[7] LC_PAPER=en_GB.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_GB.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] apeglm_1.18.0 PCAtools_2.8.0
[3] tximport_1.24.0 readr_2.1.3
[5] tzdb_0.3.0 RColorBrewer_1.1-3
[7] edgeR_3.38.4 limma_3.52.4
[9] patchwork_1.1.2 viridis_0.6.2
[11] viridisLite_0.4.0 EnhancedVolcano_1.14.0
[13] ggrepel_0.9.1 cowplot_1.1.1
[15] org.Mm.eg.db_3.15.0 AnnotationDbi_1.58.0
[17] DOSE_3.22.1 clusterProfiler_4.4.4
[19] pheatmap_1.0.12 ggplot2_3.3.6
[21] DESeq2_1.36.0 memoise_2.0.1
[23] SummarizedExperiment_1.26.1 Biobase_2.56.0
[25] MatrixGenerics_1.8.1 matrixStats_0.62.0
[27] GenomicRanges_1.48.0 GenomeInfoDb_1.32.4
[29] IRanges_2.30.0 S4Vectors_0.34.0
[31] BiocGenerics_0.42.0 Matrix_1.5-1
loaded via a namespace (and not attached):
[1] shadowtext_0.1.2 fastmatch_1.1-3
[3] plyr_1.8.7 igraph_1.3.5
[5] lazyeval_0.2.2 splines_4.2.0
[7] BiocParallel_1.30.3 digest_0.6.29
[9] yulab.utils_0.0.5 GOSemSim_2.22.0
[11] GO.db_3.15.0 fansi_1.0.3
[13] magrittr_2.0.3 ScaledMatrix_1.4.1
[15] Biostrings_2.64.1 annotate_1.74.0
[17] graphlayouts_0.8.2 bdsmatrix_1.3-6
[19] enrichplot_1.16.2 colorspace_2.0-3
[21] blob_1.2.3 dplyr_1.0.10
[23] crayon_1.5.1 RCurl_1.98-1.9
[25] jsonlite_1.8.2 scatterpie_0.1.8
[27] genefilter_1.78.0 survival_3.3-1
[29] ape_5.6-2 glue_1.6.2
[31] polyclip_1.10-0 gtable_0.3.0
[33] zlibbioc_1.42.0 XVector_0.36.0
[35] DelayedArray_0.22.0 BiocSingular_1.12.0
[37] scales_1.2.0 mvtnorm_1.1-3
[39] DBI_1.1.3 Rcpp_1.0.9
[41] emdbook_1.3.12 xtable_1.8-4
[43] gridGraphics_0.5-1 tidytree_0.4.1
[45] dqrng_0.3.0 bit_4.0.4
[47] rsvd_1.0.5 httr_1.4.4
[49] fgsea_1.22.0 ellipsis_0.3.2
[51] pkgconfig_2.0.3 XML_3.99-0.11
[53] farver_2.1.1 locfit_1.5-9.6
[55] utf8_1.2.2 labeling_0.4.2
[57] ggplotify_0.1.0 tidyselect_1.1.2
[59] rlang_1.0.4 reshape2_1.4.4
[61] munsell_0.5.0 tools_4.2.0
[63] cachem_1.0.6 downloader_0.4
[65] cli_3.3.0 generics_0.1.3
[67] RSQLite_2.2.18 stringr_1.4.0
[69] fastmap_1.1.0 ggtree_3.4.4
[71] bit64_4.0.5 tidygraph_1.2.2
[73] purrr_0.3.5 KEGGREST_1.36.3
[75] ggraph_2.0.6 nlme_3.1-158
[77] sparseMatrixStats_1.8.0 aplot_0.1.7
[79] DO.db_2.9 compiler_4.2.0
[81] png_0.1-7 treeio_1.20.2
[83] tibble_3.1.7 tweenr_2.0.2
[85] geneplotter_1.74.0 stringi_1.7.8
[87] lattice_0.20-45 vctrs_0.4.1
[89] pillar_1.8.0 lifecycle_1.0.1
[91] data.table_1.14.2 bitops_1.0-7
[93] irlba_2.3.5.1 qvalue_2.28.0
[95] R6_2.5.1 gridExtra_2.3
[97] codetools_0.2-18 MASS_7.3-58
[99] withr_2.5.0 GenomeInfoDbData_1.2.8
[101] parallel_4.2.0 hms_1.1.2
[103] grid_4.2.0 ggfun_0.0.7
[105] beachmat_2.12.0 coda_0.19-4
[107] tidyr_1.2.1 DelayedMatrixStats_1.18.1
[109] bbmle_1.0.25 ggforce_0.4.1
[111] numDeriv_2016.8-1.1

Thanks, makes sense!