Entering edit mode

Hi,
I find the plotUMAP for reconstructed value after fastMNN is not readable.
For example, here is my interesting gene in replicate 1
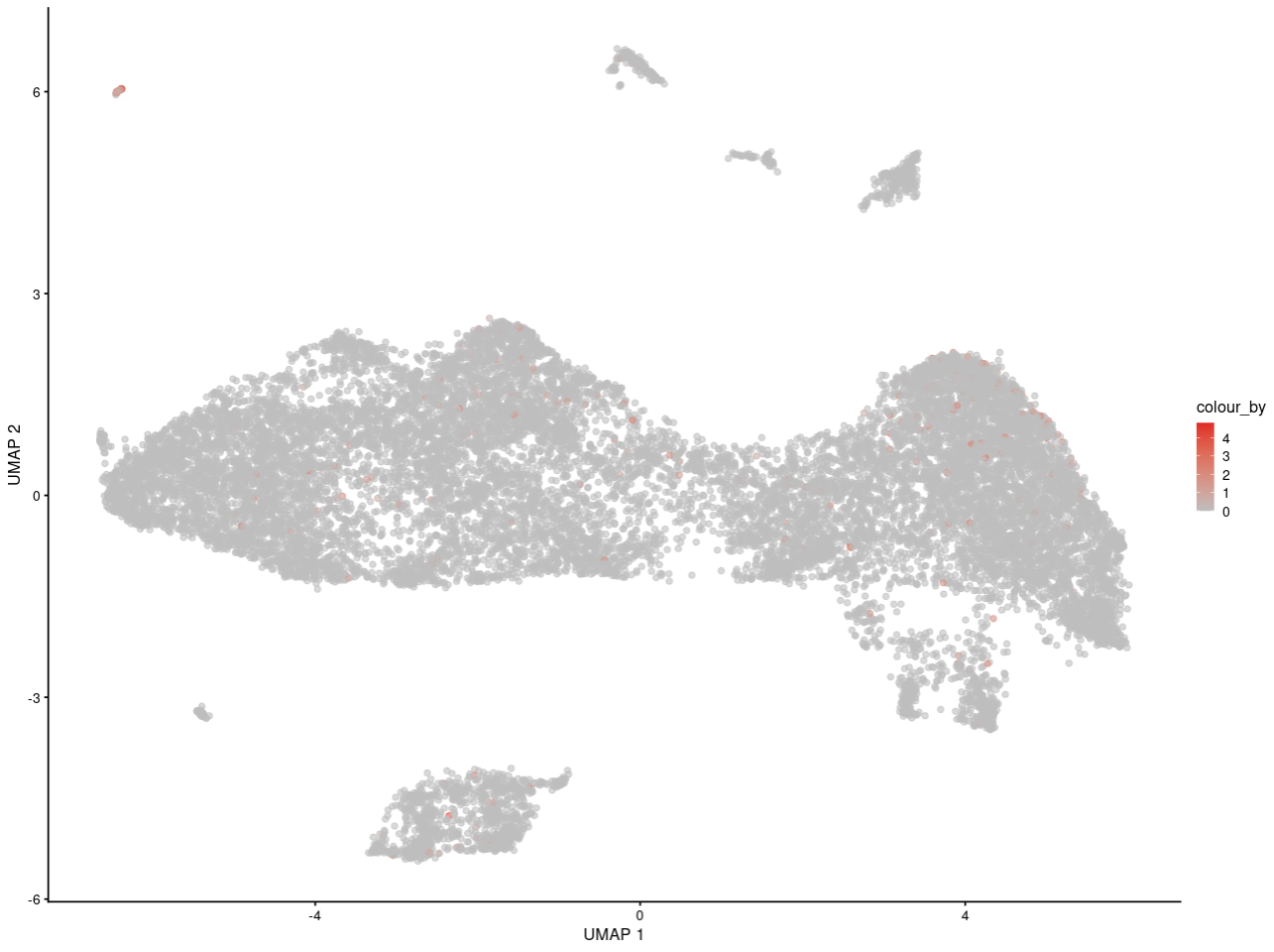
and here is in replicate 2
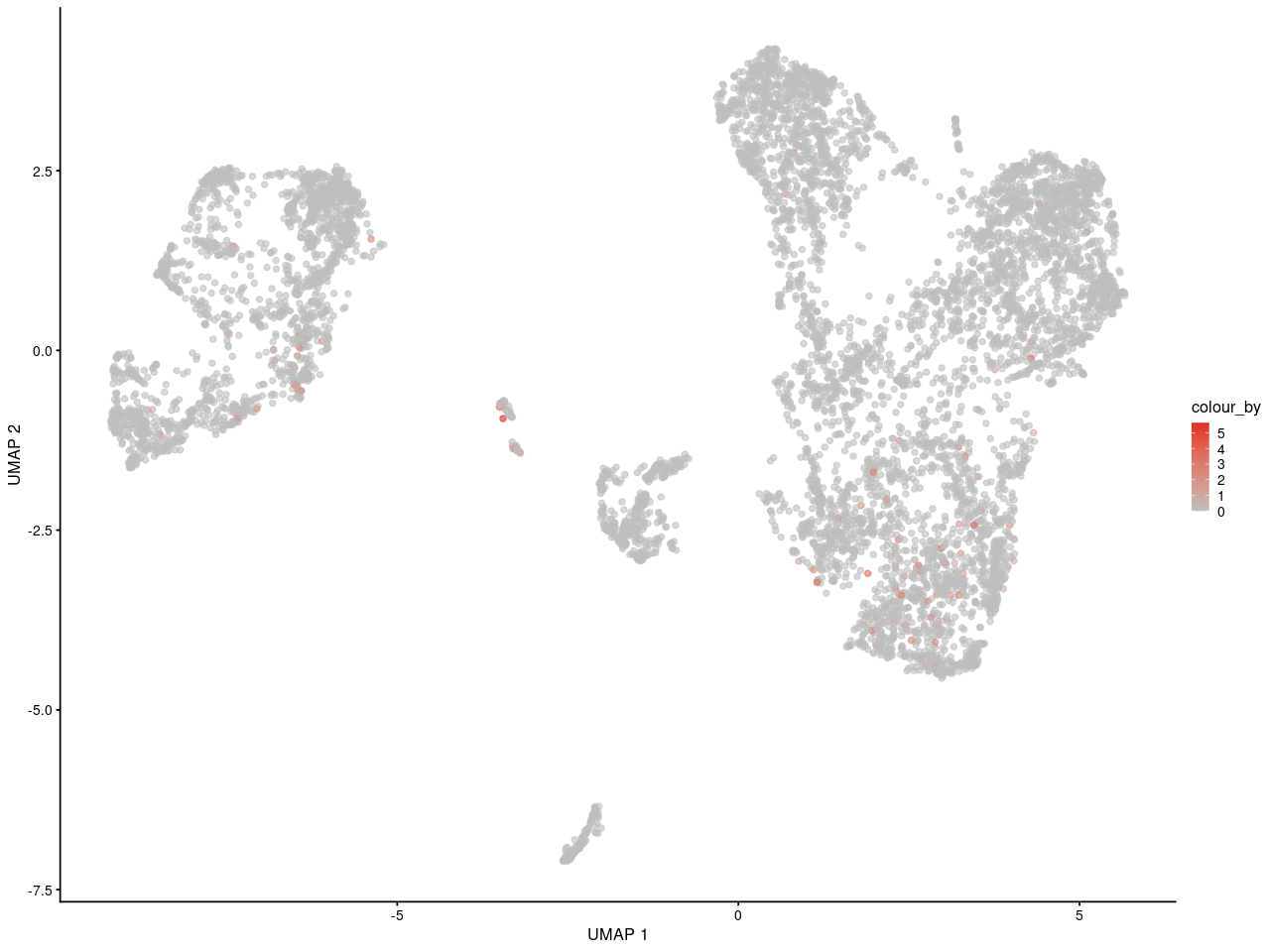
As you can see, I can see the gene may express in specific some cells. And for people who is not familar with scRNA-seq, they can get the same information as me.
And here is the vilion plot
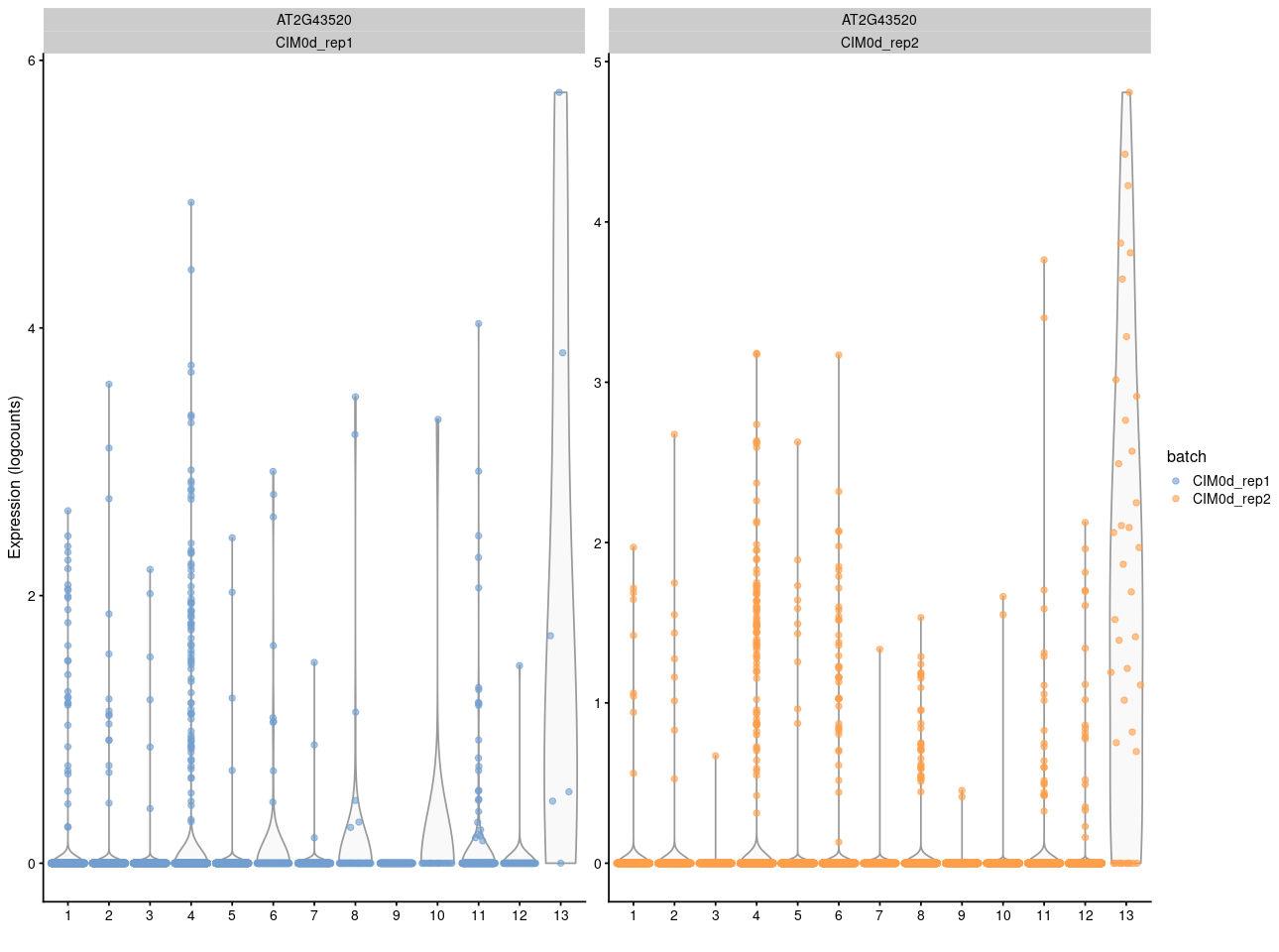
But if I want to show the gene in merged UMAP after fastMNN using the reconstructed value, it seems for other people that this gene is expressing eveywhere while some cells express high and some cells express low count.
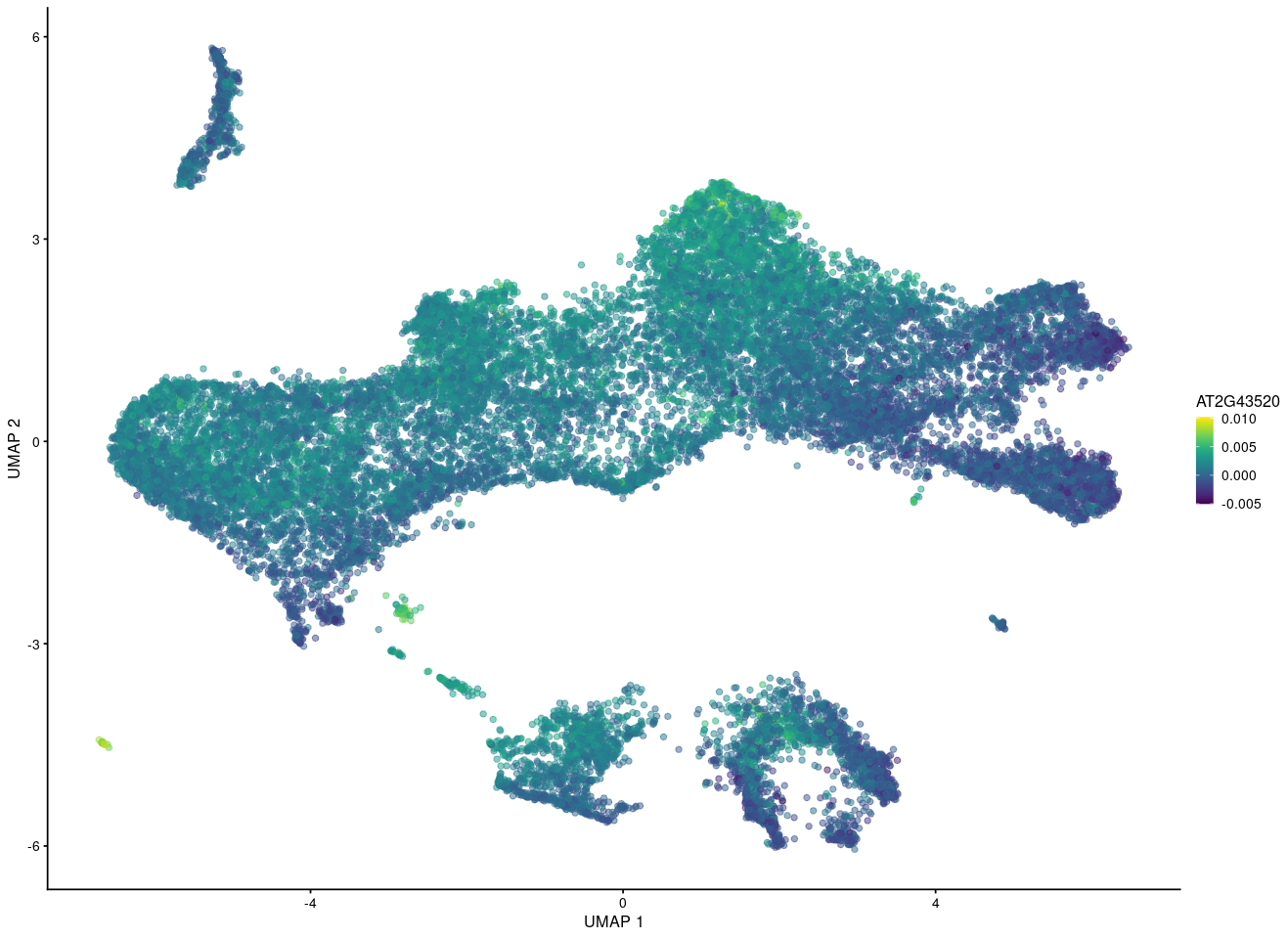
So I am wondering whether some one can give me some advices about it.
Best wishes
Guandong Shang

What's the last plot from? The scale on the colour axis seems to have a very small range, which makes it seem like this may not actually represent large differences in expression between "high" and "low" groups. I imagine it would be possible to scale the values shown in your plot to make something similar, though I obviously would not advise you to do that.
However it's really hard to say what's "wrong" here without knowing further details.
Hi, thanks for your reply.
according to the OSCA manual about fastMNN
http://bioconductor.org/books/3.15/OSCA.multisample/integrating-datasets.html#mnn-correction
I use this reconstructed value to show my gene expression on UMAP plot. And here is my summary about my gene
By the way, it seems that Seurat also have this problem for intergated scRNA-seq expression on UMAP according to this issue. They maybe use the
min.cutoffparameter to drop these "non-expression" value?