Entering edit mode

Hi everyone!
I am unsure what is causing my DMRfinder R program to not work. I did not use the DMRfinder pileup step (using bismark) because I wanted to test my own pileups. Therefore, I reformatted existing bedgraph files to have the following format:
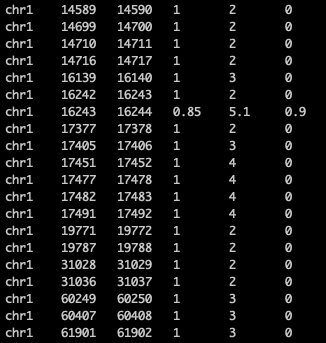
I then got the following combined bed file from running two bed samples:
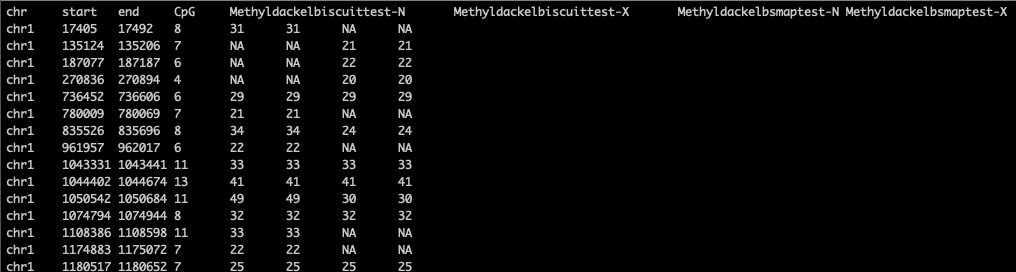
Here is the command line I ran while I was cd'd into the folder that all these files are/would be located:
Rscript /pathtocode/DMRfinderinst/DMRfinder-master/findDMRs.r -i both.combined.bed -o DMRsFound.csv Methyldackelbiscuittest Methyldackelbsmaptest -n Methyldackelbiscuittest,Methyldackelbsmaptest -d 0 -p 1
My output was as follows:
There were 23 warnings (use warnings() to see them)
Error in if (substr(level[i], 1, 3) == "chr") { :
argument is of length zero
Execution halted
I admit I am not proficient at R myself to troubleshoot this issue. At first I thought this was because some of my chr outputs were formatted as satellite chromosomes, but this was after I subsetted out the non chr 1:22, chrX, chrY chromosomes.
Any insight would be helpful, thank you!
Hi Claire!
Did you find any solution? I have the same problem :`)