Entering edit mode

Hi,
Library size is a source of unwanted variation in my data (dataset of healthy vs disease samples) because I can see in the pca plot that samples are separated to some degree by library size. Can sva remove the effect that library size has on my data or is another package better suited for this? To be clear: I'm not trying to remove batch effects.
Thanks!

Hi thanks for answering! This is the code that I want to use:
However, I'm unsure about the mod0 line which should only include the "adjustment" variables, so that would be libsize I think? I'm pretty sure I'm making a mistake with the svseq line because I don't want to add any surrogate variables, just remove lib.size as a confounder, but I don't know how to write this down. I'd really appreciate your help!
The plots aren't really the problem, it's just the code that is giving me great difficulties. Thanks again!
You are generating surrogate variables, but you never use them. Also mod0 should include all the adjustment variables, which includes sex (which is almost always included as a nuisance variable rather than a parameter of interest). Also, if you are fitting lib.size as part of your model, then the surrogate variables won't adjust for library size unless there are differences in variability due to library size (which is not a completely unexpected result, particularly if you have widely divergent library sizes).
Also also, you don't want to use lib.size as a batch argument in
removeBatchEffect. The expectation for a batch is that it is a factor, whereas lib.size is continuous. One of the first things that happens to the batch argument is conversion to factor. If you use a continuous variable you will end up with N factor levels (where N is the number of samples), which is not what you want to do. The covariates argument is what you use for continuous variables. And if you want to adjust for the surrogate variables you have tocbindthe lib.size and svseq$sv together and pass that toremoveBatchEffect.Thanks for the help! I have now changed libsize to a continous variable (it was a dichotomous variable: high/low at first). I should use the pre-normalized counts for this right? My code is now as follows:
This gives me the following plot: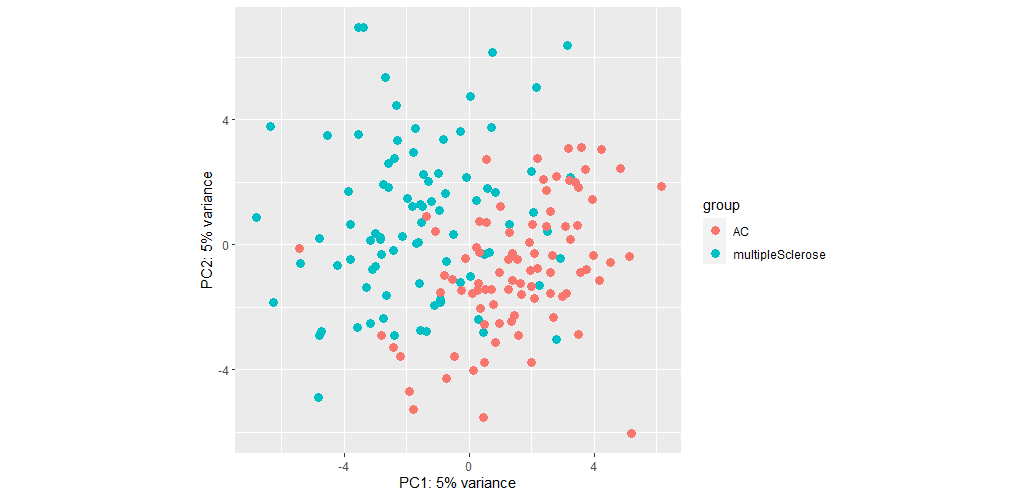
The seperation isn't bad but PC values are low. When I do not remove SV, the PC values increase but separation worsens: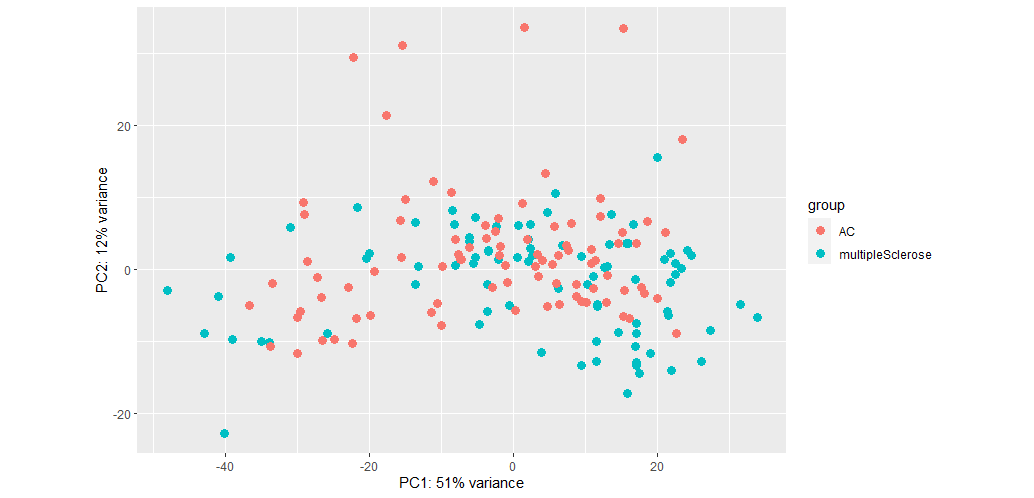
So would it be fair to conclude that batch effect is more important than biological effects for my dataset?
Do you have batches? If you have known batches, people usually directly account for them as part of the linear model rather than doing something indirect like computing surrogate variables.
I actually don't have known batches (I'm using data from another researcher from some time ago who does not have this information anymore). Do you think that my code lines look ok in general? On another note: the sva code line
Gives me the following error:
Error in solve.default(t(mod) %*% mod) : system is computationally singular: reciprocal condition number = 1.32097e-16I have found that you can get this by two covariates (patient group and libsize in my case, I presume) being highly colinear or because the number of cases is approximately as large as the number of covariates. These are both not true for my dataset I think (case number is 174, covariate number is 2). Do you have any idea if something else could cause this error?And would it be possible to remove confounders using ruvseq and limma's remove batch effect function?
Yes, that is another method you could use.