
I have encountered something really confusing when I did my LRT analyses. I'm interested in gene P's effect on cellular immune reactions. In experiments, we treated the WT and P KO cells with either normal media (control), or drug A, or drug B to induce immune responses. For each treatment, we treat the cells for 6 hours and 24 hours.
In these experiments, we find a change in metabolites in WT cells treated with drug A for 6 hours, and also in WT cells treated with drug B for 24 hours. We didn't see this metabolic change in P KO cells. To understand what is going on, we did RNAseq experiments on these WT and KO cells.
Biological question: for a given treatment, which genes are differentially expressed in WT, that do not change expression in P KO (or change differently). Basically what impact does P KO have the transcriptional pattern across treatment (untreated/media, 6h, 24h). We expect some genes given the differences in metabolites observed (described above).
If we divide the data by treatment, my metadata looks like this:
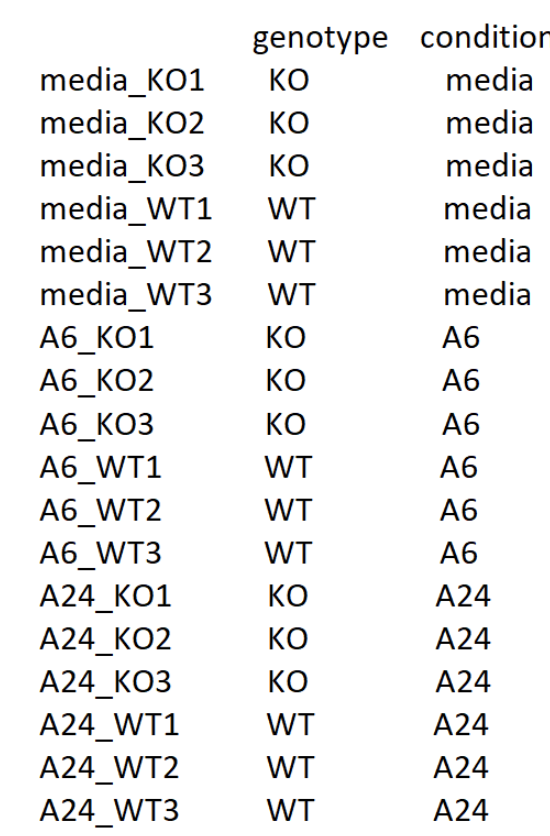
For this purpose, I used the LRT with the interaction term to understand gene P’s effect on the treatment. But when I do this, all the genes show padj =1, which doesn’t make sense. So I wonder if there is anything wrong in my code? Or is there other better way to analyse this data?
dds_geno <- DESeqDataSetFromMatrix(data, colData = meta_A, design = ~ genotype + treatment + genotype:treatment)
dds_geno_lrt <- DESeq(dds_geno, test="LRT", reduced = ~ genotype + treatment)
Many thanks in advance!
Meeta
