
If I have 2 different codesets from nanostring, how should I run "RUV_total" function?
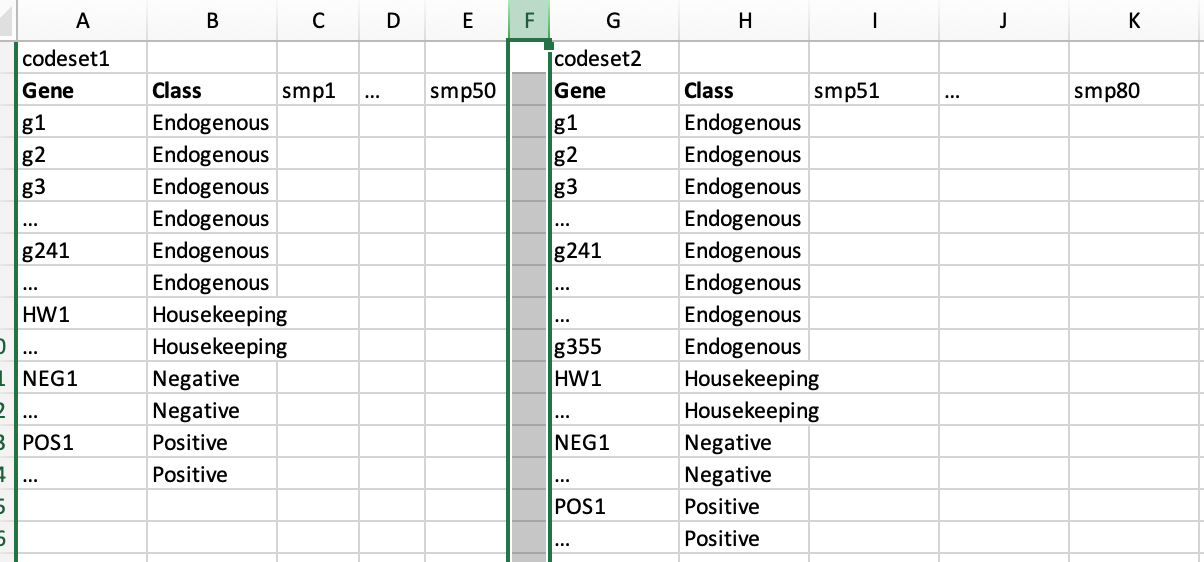
"Different codesets" means the dimension of 2 or more sets is different, such as below. There are a set of Endogenous genes are in common between the 2 sets. The house keeping (hk), negative, and positive genes are same for 2 sets. I have 2 ways in my mind to run "RUV_total" function. But, I have no idea which one is more reasonable, or better ideas?
First, I can merge the codesets, and then run "RUV_total" function on the merged one. For example, there are 241 Endogenous + 20 (hk, neg, pos) genes for 80 samples after merging. In this way, the genes which are not in common were removed, my concern is that if the removal of uncommon genes will have effect on ruv normalization.
Second, I can do normalization on codeset1 and 2, respectively. Then, the RUV factors can be combined to be pData and passed to DESeq2. In this case, the "countData" is still same as the above way.
Not sure if the "codeset size" (like the library size for RNAseq) has effect on ruv normalization or counts(dds, normalized=TRUE) function.
Any ideas or suggestions would be appreciated!
I don't have an answer from the DESeq2 side, I'll leave this for the RUV devels.
I played in more details. @Michael love, do you have any comments about my play? I also copied this thread to GitHub (CBCS_normalization). Thanks a lot!
I calculated the W_1 for those 2 ways, respectively. The plot is as below. "preMer_w1" means, RUV_total was run on each of codeset individually. "postMer_w1" means, RUV_total was run on the merging codesets. In this case, only the common genes were kept in the merging data frame. The W_1 factor is different, which is within my expectation because different datasets were used, although I have no idea if some correlation was expected between them.
Then, I passed the W_1 to DESeq2 and got that the normalized counts are exactly same (RLE plots were exactly same), which is surprising to me.
The codes is as blow:
counts(dds, normalized=TRUE)scales to account for size factor (sequencing depth) but doesn't remove variance associated with the design variables.oh, I see the design variables are not used when estimating the size factors, and counts(dds, normalized=TRUE) is providing counts scaled by size or normalization factors.
How could I get the normalized counts accounting for both size factors and design variables? So, I can use those normalized counts for RLE plots. I tried to google, but didn't find an answer.
In the DESeq2 vignette, we suggest you can use the VST then followed by regressing out batch variables.
So, the code should be like this in the vignette as below. But, I have no idea what is the batch variable in my case. If it will be the different codeset, I think I need to combine pData(RUVnorm.dat_codeset262_375_post$set) and pData_preMerge, and set a new variable of "batch"? Then, run
vignette codes:
It's up to you what you want to regress out (and for what analyses/purposes) I was just pointing to code that allows for "...accounting for both size factors and design variables".