
Hi Aaron.
I have Time-series bulk data (with two biological replicates for each time) and now I wish to use it to annotate my single-cell data. My Single-cell data contains two samples, One control and one drug-treated.
Your vignette is quite straight forward but can you tell me how I combine two biological replicates per time point into one column so that I have one column for each time point and use it for assigning labels. Will log normalizing both replicates and then take average per gene per time point work?
Update1: I actually did it as I mentioned above and used the following code. My cells are highly synchronized at 10 hours post-infection and I was using Bulk RNASeq to confirm the same using SingleR. But I am getting score 1 for all the time points. However, the final label assigned is correct i.e 10hpi. Weird though that the heatmap is all blue not yellow!!
library(SingleR)
library(Seurat)
library(dplyr)
c <- read.csv("../time_series_data/counts.csv", header = T, row.names = 1)
> c %>% head()
T0_rep1 T0_rep2 T2_rep1 T2_rep2 T4_rep1 T4_rep2 T6_rep1
Gene1 46 48 202 298 357 428 125
Gene2 130 137 693 844 1713 1504 630
Gene3 0 0 0 0 1 0 0
Gene4 1 1 1 0 0 1 1
Gene5 412 524 994 1569 1959 2095 771
Gene6 16909 16966 16490 23660 30364 30484 10008
meta <- read.csv("../time_series_data/meta.csv", header = T)
> meta %>% head()
Sample_id Prefix Replicate Time timetag
T0_rep1 T0_rep1 T0 rep1 4 4hpi
T0_rep2 T0_rep2 T0 rep2 4 4hpi
T2_rep1 T2_rep1 T2 rep1 6 6hpi
T2_rep2 T2_rep2 T2 rep2 6 6hpi
T4_rep1 T4_rep1 T4 rep1 8 8hpi
T4_rep2 T4_rep2 T4 rep2 8 8hpi
rownames(meta) <- meta$Sample_id
bulk <- CreateSeuratObject(counts = c, project = "Time-Series")
bulk$Sample <- "T-series"
bulk$batch <- "T-series"
bulk$batch2 <- "T-series"
bulk@meta.data <- cbind(bulk@meta.data,meta)
bulk <- NormalizeData(bulk)
prefix <- unique(meta$Prefix)
mat.temp <- data.frame(GetAssayData(bulk, slot="data"))
l <- list()
biorepavg <- lapply(1:length(prefix),function(x){
l[[x]] <- mat.temp[,grep(prefix[x], colnames(mat.temp))] %>% rowMeans()
})
df <- data.frame(sapply(biorepavg,c))
colnames(df) <- prefix
newmeta <- meta[,c(2,4,5)] %>% unique() %>% `rownames<-`(.$Prefix)
> newmeta %>% head
Prefix Time timetag
T0 T0 4 4hpi
T2 T2 6 6hpi
T4 T4 8 8hpi
T6 T6 10 10hpi
T8 T8 12 12hpi
T10 T10 14 14hpi
tseries <- SingleCellExperiment(list(logcounts=df),colData=DataFrame(label=newmeta))
> pred.10nt.ts <- SingleR(test=p10NT.sce, ref=tseries, labels=tseries$label.timetag, de.method="wilcox",de.n=20)
> pred.10nt.ts$scores %>% head()
10hpi 12hpi 14hpi 16hpi 18hpi 20hpi 22hpi 24hpi 26hpi 28hpi 2hpi
[1,] 1 1 1 1 1 1 1 1 1 1 1
[2,] 1 1 1 1 1 1 1 1 1 1 1
[3,] 1 1 1 1 1 1 1 1 1 1 1
[4,] 1 1 1 1 1 1 1 1 1 1 1
[5,] 1 1 1 1 1 1 1 1 1 1 1
[6,] 1 1 1 1 1 1 1 1 1 1 1
30hpi 32hpi 34hpi 36hpi 38hpi 40hpi 42hpi 44hpi 4hpi 6hpi 8hpi
[1,] 1 1 1 1 1 1 1 1 1 1 1
[2,] 1 1 1 1 1 1 1 1 1 1 1
[3,] 1 1 1 1 1 1 1 1 1 1 1
[4,] 1 1 1 1 1 1 1 1 1 1 1
[5,] 1 1 1 1 1 1 1 1 1 1 1
[6,] 1 1 1 1 1 1 1 1 1 1 1
> pred.10nt.ts$labels %>% head()
[1] "10hpi" "10hpi" "10hpi" "10hpi" "10hpi" "10hpi"


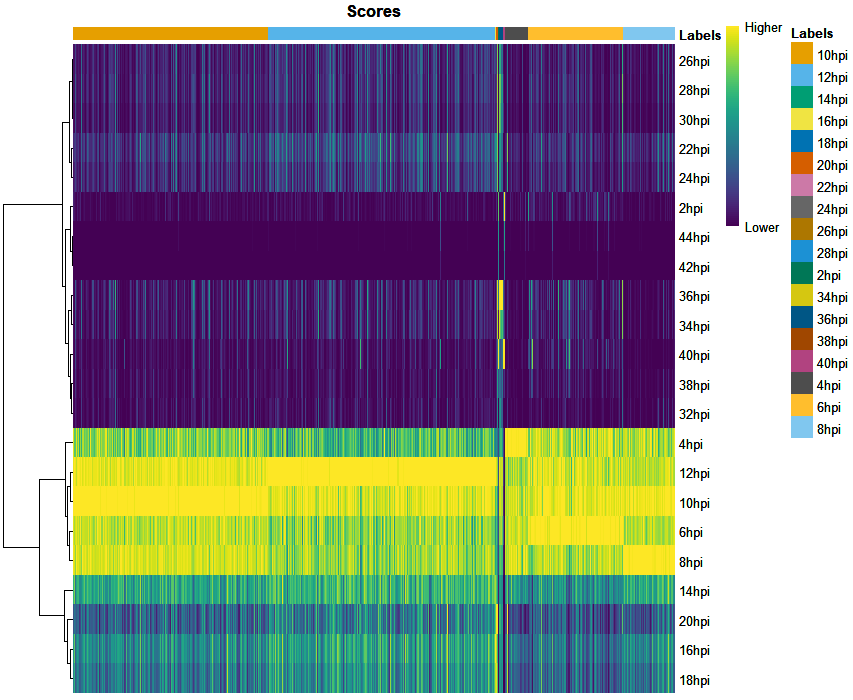
UPDATE 2: Repeated the analysis after removal of ribosomal protein genes from the bulk. But still getting the same results. Plotted the Correlation heatmap to see how similar the transcriptional profile are between time points using
CHETAHfunctionCorrelateReference()as shown below.Since the transcriptional profile of 10hpi highly correlates with 12 and 14hpi, I tried subsetting the Bulk data keeping samples with 4 hrs difference such as
2hpi,6hpi,10hpi,14hpi .....but still, I observe no changes in the results.