
Hi everyone,
I have a question about the GSVA package, In particular about the results with different methods parameter value ("ssGSEA" and "GSVA" ). In my case I have 5 sample and I want to test the enrichment of 2 genesets on them. The step of my analysis was the following:
- produce a raw counts matrix for BAM files
- normalise the raw counts (DESeq2)
- transform the normalised counts via regularised log
- Run GSVA
I've performed the gsva() 2 times with same parameter except for the method; one time i used ssGSEA and for the other GSVA, the results i've obtained led to opposite conclusions (regarding the enrichment of the two genesets). I've attached the plot showing the obtained scores
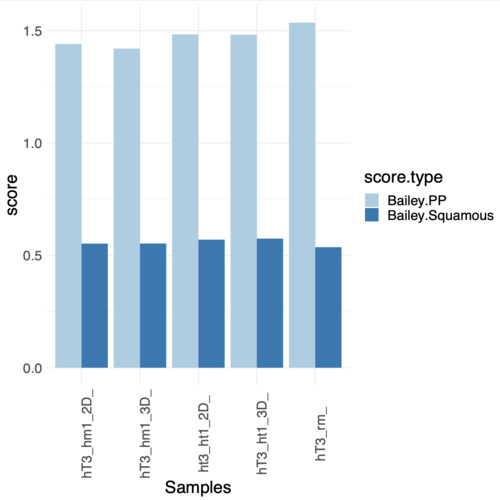
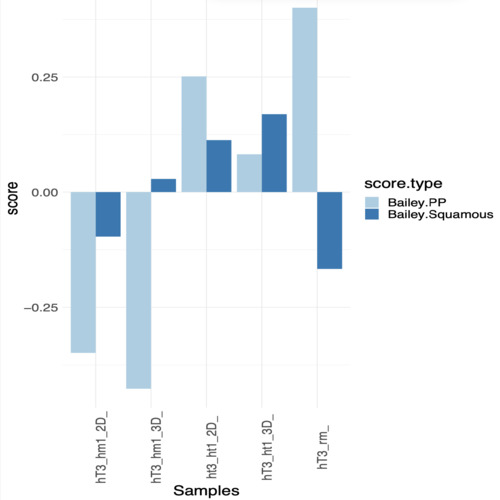
My question is, it's possible that the two methods gives me such opposite results when described as "similar" in the help page? I've read the reference paper trying to understand better which could be the source of the variation but i think i'm loosing something (or more), it's probably due to the gene ranking step? even if it's due to that or from other aspects of the computation i didn't expect, from what i know, such a difference in results. If someone can fill my gap in knowledge for big it is and give me some insights/explanations on the argument and results understanding it would be awesome. Let me know if more information are needed from me on the problem.

Hi, Thanks for the reply, i looked at the genes expressions data and i have an idea which result have more sense. At the same time i'm posting here data and code, if you have time to look at that, it will be awesome.
here the data: https://univr-my.sharepoint.com/:u:/g/personal/davide_pasini_univr_it/EaPXg7f2LrFLlXId8JoKSSkBKhhUwoeqTBMrjdW4MW_n6Q?e=fmKA1G[ link]1
here a trimmed version of the code i used, with the main workflow:
ok, from what i see both results, GSVA and ssGSEA are quite proportional to each other, if you rename the objects that get the result in the last two calls from the previous code, to
dseqobj_gsvaanddseqobj_ssgsea, respectively, and you plot them doing:you should see what i'm talking about, they are linearly correlated with each other with rho about 0.95. The difference is in the scale of values that you get from each method. In any case, you say you have an idea which result make more sense to you, so that should help you decide how do you want to do your analysis.
As a side note, I notice that after filtering lowly expressed genes, you still keep about 24,000, this is I think way too many because usually only about the half are expressed in a given experimental condition, tissue, etc. If you look at the distribution of average expression values per gene by doing:
You will still see two well separated modes, the one on the left is made of genes that are probably not reliably expressed. I'd suggest to remove those.