Entering edit mode

Hello all,
I'm using ATACseqQC package for analyzing my post-sorted bam file of ATAC-seq (from mice).
I found my heatmap was only half shown whatever I tried. Please see the code below.
# for mouse
library(ATACseqQC)
library(ChIPpeakAnno)
library(MotifDb)
library(GenomicAlignments)
library(Rsamtools)
library(GenomicScores)
library(BSgenome.Mmusculus.UCSC.mm10)
library(TxDb.Mmusculus.UCSC.mm10.knownGene)
mm10_gscore <- getGScores("phastCons60way.UCSC.mm10")
txs <- txs[seqnames(txs) %in% c("chr1", "chr2", "chr3", "chr4", "chr5", "chr6", "chr7", "chr8", "chr9", "chr10", "chr11", "chr12", "chr13", "chr14", "chr15", "chr16", "chr17", "chr18", "chr19", "chrX", "chrY")]
genome <- Mmusculus
objs <- splitGAlignmentsByCut(obj=gal1, txs=txs, genome=genome, outPath="D:/HYJ/splited_Cracd_KO1/")
bamfiles <- file.path("D:/HYJ/splited_Cracd_KO1/", c("NucleosomeFree.bam", "mononucleosome.bam", "dinucleosome.bam", "trinucleosome.bam"))
TSS <- promoters(txs, upstream=0, downstream=1)
TSS <- unique(TSS)
librarySize <- estLibSize(bamfiles)
librarySize
D:/HYJ/splited_Cracd_KO1//NucleosomeFree.bam D:/HYJ/splited_Cracd_KO1//mononucleosome.bam
7552685 1484090
D:/HYJ/splited_Cracd_KO1//dinucleosome.bam D:/HYJ/splited_Cracd_KO1//trinucleosome.bam
1497497 0
NTILE <- 101
dws <- ups <- 1010
sigs <- enrichedFragments(gal=objs[c("NucleosomeFree", "mononucleosome", "dinucleosome", "trinucleosome")], TSS=TSS, librarySize=librarySize, upstream = ups, downstream = dws, TSS.filter=0.5, seqlev = paste0("chr", c(1:19, "X", "Y")), n.tile = NTILE)
sigs.log2 <- lapply(sigs, function(.ele) log2(.ele+1))
featureAlignedHeatmap(cvglists=sigs.log2, feature.gr=reCenterPeaks(peaks=TSS, width=ups+dws), upstream = ups, downstream = dws, zeroAt=0.5, n.tile=NTILE)
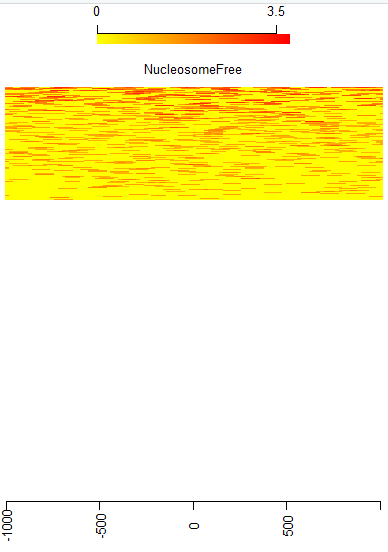
I think the reason may be the NA values in sigs. And the NA values may be caused by the not well-aligned bam files because there is no conservation=mm10_gscore in objs <- splitGAlignmentsByCut(obj=gal1, txs=txs, genome=genome, outPath="D:/HYJ/splited_Cracd_KO1/"). However, if I add conservation=mm10_gscore, the splitGAlignmentsByCut() will generate Error: subscript contains invalid names.
sigs
$NucleosomeFree
[,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10] [,11]
[1,] NA NA NA NA NA NA NA NA NA NA NA
[2,] 1.5850972 1.5850972 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
[3,] 0.0000000 0.0000000 0.000000 0.000000 0.000000 0.000000 0.000000 2.451784 2.451784 2.451784 2.451784
[4,] 0.0000000 0.0000000 0.000000 0.000000 0.000000 1.585097 1.585097 1.585097 1.585097 1.585097 0.000000
[5,] 0.0000000 1.2258919 1.225892 1.225892 1.225892 1.225892 0.000000 0.000000 0.000000 0.000000 0.000000
[6,] NA NA NA NA NA NA NA NA NA NA NA
[7,] NA NA NA NA NA NA NA NA NA NA NA
[8,] 0.0000000 0.0000000 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
[9,] NA NA NA NA NA NA NA NA NA NA NA
It is appreciated if anyone could share some opinions or tips on this issue. Thanks! Best, YJ