
Hi, there:
I have a question about how plotSA shall look like for good or bad data, any example of good or bad data shall look like in plotSA regarding whether the data is OK for limma analysis or not?
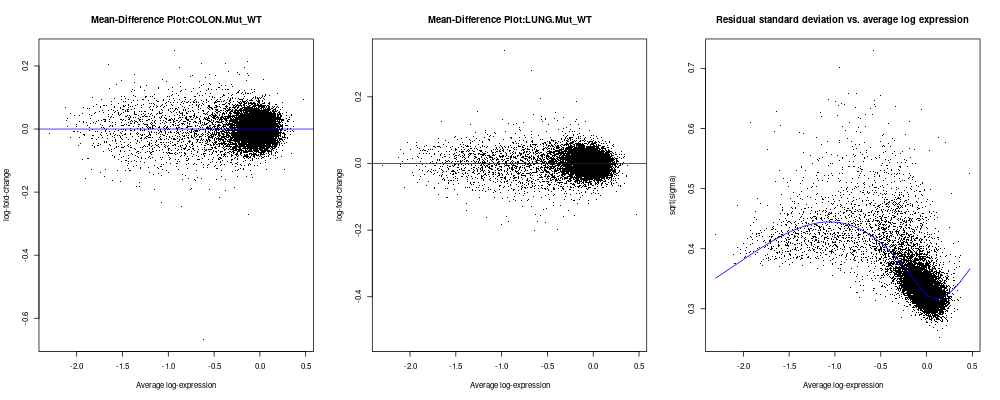
I indeed looked through web or the user guide of limma or the limma method paper, I could not find example plots of good or bad plotSA plots shall look like. So hereby I wonder if one can offer examples on what kind of plotSA plots shall look like or what criteria to determine regarding to whether the data is OK for limma analysis or not. Since if plotSA plot can be a diagnosis tool to determine if limma can be applied to the data as discussed initially and partially at URL: old discussion, it is better to know how and what is OK and what is not OK look like. I attached example of my plotSA plot here with also plotMD plots for reference here and code is below. The plotSA plot for my data look kind of bent and skewed, although plotMD look great.
Thanks in advance! Best
Ming
Code should be placed in three backticks as shown below
> show(mydata[1:5,1:3]);
COLON.Mut.LS513 COLON.Mut.CL11 COLON.Mut.LS1034
A1BG..1. 0.009157821 -0.001640884 -0.05054882
A1CF..29974. -0.021848711 0.036079619 0.05448993
A2M..2. 0.099182798 0.105247983 0.04387373
A2ML1..144568. 0.107724902 0.043404811 0.02711932
A3GALT2..127550. -0.045207710 -0.146916234 -0.37110402
> show(tar[1:5,c(8,7,3)])
TypesLines RAS Types
COLON.Mut.LS513 COLON.Mut.LS513 Mut COLON
COLON.Mut.CL11 COLON.Mut.CL11 Mut COLON
COLON.Mut.LS1034 COLON.Mut.LS1034 Mut COLON
COLON.Mut.SNU1033 COLON.Mut.SNU1033 Mut COLON
COLON.Mut.COLO678 COLON.Mut.COLO678 Mut COLON
eset <- ExpressionSet(assayData = as.matrix(mydata))
group1<-paste(tar$Types,tar$RAS,sep=".");
group<-factor(group1,levels=c("COLON.Mut","COLON.WT","LUNG.Mut","LUNG.WT"));
design <- model.matrix(~0+group)
colnames(design) <- levels(group)
fit <- lmFit(eset, design)
> show(fit)
An object of class "MArrayLM"
$coefficients
COLON.Mut COLON.WT LUNG.Mut LUNG.WT
A1BG..1. -0.006184935 -0.01589437 -0.01650645 -0.03327325
A1CF..29974. -0.036653106 -0.05470048 -0.03705100 -0.05318098
A2M..2. 0.059910009 0.05123221 0.08906358 0.07615483
A2ML1..144568. 0.062222203 0.12934582 0.10457048 0.12544309
A3GALT2..127550. -0.142014114 -0.12313766 -0.15668482 -0.17035788
17640 more rows ...
.......
expName<-paste("Test_PlotSA_LungColon_V2", "",sep="");;
pngF<-paste(outPath,paste(expName,".png",sep=""),sep="/");
png(file= pngF, width = 1000, height = 400);
par(mfrow=c(1, 3))
cont.matrix<-makeContrasts(COLON.Mut_WT=COLON.Mut-COLON.WT, LUNG.Mut_WT=LUNG.Mut-LUNG.WT,levels=design);
fit2 <- contrasts.fit(fit, cont.matrix)
fit3 <- eBayes(fit2)
> show(fit3)
An object of class "MArrayLM"
$coefficients
Contrasts
COLON.Mut_WT LUNG.Mut_WT
A1BG..1. 0.009709434 0.01676680
A1CF..29974. 0.018047372 0.01612999
A2M..2. 0.008677799 0.01290875
A2ML1..144568. -0.067123620 -0.02087261
A3GALT2..127550. -0.018876454 0.01367307
17640 more rows ...
.......
plotMD(fit3, coef=1,main=paste0("Mean-Difference Plot:", colnames(fit3$coefficients)[1]));
abline(0,0,col="blue")
plotMD(fit3, coef=2,main=paste0("Mean-Difference Plot:", colnames(fit3$coefficients)[2]));
abline(0,0,col="blue")
plotSA(fit3, main="Residual standard deviation vs. average log expression")
dev.off()
I atttached the image of the created plots here for your view
> sessionInfo()
R version 4.0.2 (2020-06-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS: /mnt/nasapps/development/R/4.0.2/lib64/R/lib/libRblas.so
LAPACK: /mnt/nasapps/development/R/4.0.2/lib64/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] calibrate_1.7.7 MASS_7.3-54 Biobase_2.50.0
[4] BiocGenerics_0.38.0 limma_3.46.0 reshape_0.8.8
[7] reshape2_1.4.4 ggrepel_0.9.1 ggplot2_3.3.5
loaded via a namespace (and not attached):
[1] Rcpp_1.0.7 pillar_1.6.2 compiler_4.0.2 plyr_1.8.6
[5] tools_4.0.2 digest_0.6.27 lifecycle_1.0.0 tibble_3.1.3
[9] gtable_0.3.0 pkgconfig_2.0.3 rlang_0.4.11 DBI_1.1.1
[13] withr_2.4.2 dplyr_1.0.7 stringr_1.4.0 generics_0.1.0
[17] vctrs_0.3.8 grid_4.0.2 tidyselect_1.1.1 glue_1.4.2
[21] R6_2.5.0 fansi_0.5.0 farver_2.1.0 purrr_0.3.4
[25] magrittr_2.0.1 scales_1.1.1 ellipsis_0.3.2 assertthat_0.2.1
[29] colorspace_2.0-2 labeling_0.4.2 utf8_1.2.2 stringi_1.7.3
[33] munsell_0.5.0 crayon_1.4.1

Dr, Smyth:
Thanks so much for taking time respond to my questions. I apologize about being persistent and even a bit stubborn to keep asking seemingly a same question. One reason is: probably in contrast to what you might have thought, indeed, I was so impressed and even a bit overwhelmed by how well limma has performed in my datasets as disucssed to reflect what exatcly I thought in terms of making great biological sense of the data even more than expected, to the extent almost too good to be true. Of course, as limma paper stated, I understood limma borrows information across genes and leverages information from the within-group replicates to obtain much higher significance levels than the native t-test etc, which is why we have limma method developed by you as one of the greatest methods I ever experienced. I even did lots of simulations to compare randomly selected contrasts with million trials of limma analyses to validate my observations, which all come great. This left me just one big question as I kept asking you: how to evaluate and what kind of data limma method can be applied for? and is limma applicable to my dataset given the plotSA I got with bent and skewed curve, which was asked by my colleagues. I do not know the prerequisite of limma and did not see examples of plotSA in user guide, which is why I kept asking. Now thank you for being critical on our data quality, but it is cell assay-based high throughput data, which has been processed in very complicated procedures for experimental procedure-based reasons, and for other purposes as well as proprietary reasons, and is not under my control. I just happened to think about using limma to deal with the processed data for the questions that I am asking about. Another reason is from this experience and past ones, I trully appreciate the power of limma method and its wide potential applications in many types of high-thoourghput datasets, and that is why I wish to have a way to judge on my own to make sure limma can be applicable to many potential datasets not just common genomic array or q-PCR data etc. Thank you for your last point, which seems giving new cues in terms of the SA plot and sounds encouraging. Your 4th point mentioned a possible way of improving the data by stabilizing the variance properly, which we have touched a bit in earlier discussion. Do you mind suggesting an actual way or R package/function that I can try that can optimize the data as input for limma? Again, I am very grateful for all your kind help and certainly will cite limma and acknowldege your help if my this work can be ever published later. Thank you!
Can I suggest a way to optimize your data? No I can't. Data preprocessing is technology specific. No one can tell you how to preprocess your data because we don't have examples of data from your assay technology. We don't even know what your technology is. Please read the last sentence of my point 2 above. You have already explained that the data is proprietary and not under you control so it is very difficult indeed to understand what you are expecting me to suggest.
Thanks for the input! What I am asking is not to optimize the whole data preprocessing, since it has been done and it is techonology dependent, and no need and would be more issues to do anything on that side. I am just wondering about any potential way of stabilizing the variance of processed data properly as observed in plotSA and as you suggested.
I have no idea how to improve your data. I've never seen anything similar to it. If you want info about standard variance stabiliizing transformations, do a Google search (as I suggested 4 months ago). But I have no expectation that any of the standard transformations will be appropriate for your data. The need for variance stabilization generally needs to be incorporated at early stages of the data processing steps. It is not always possible to fix the data by tacking on a transformation at the end.
It seems from your comments that you already have perfectly satisfactory results and that the limma package has performed correctly. If you still need more assistance, the logical step would be to collaborate with a statistical bioinformatician at your own institution.
thank you for your great help!