Entering edit mode

Hi all and thank you for your help in advance.
I am plotting the results of DESeq2 output using Enhanced Volcano package. I also want to plot only 20 gene labels.
My result table:
> head(resC3)
row baseMean log2FoldChange lfcSE stat pvalue padj symbol geneName
1 ENSMUSG00000102693 1.5184259 -0.7800748 3.238902 -0.24084540 0.8096749 0.9920008 <NA> <NA>
2 ENSMUSG00000051951 4.4347206 0.7240531 1.907310 0.37962014 0.7042274 0.9896822 Xkr4 X-linked Kx blood group related 4
3 ENSMUSG00000102851 0.7409902 -3.2522220 3.440876 -0.94517258 0.3445708 0.9896822 <NA> <NA>
4 ENSMUSG00000103377 2.1128707 0.1741636 3.016754 0.05773211 0.9539620 0.9965102 <NA> <NA>
5 ENSMUSG00000104017 2.3202905 1.3844931 3.316122 0.41750370 0.6763100 0.9896822 <NA> <NA>
6 ENSMUSG00000103025 0.6405565 0.8586579 3.443174 0.24937975 0.8030670 0.9908850 <NA> <NA>
Rearrange the color:
keyvals.colour <- ifelse(
resC3$log2FoldChange < 0 & resC3$padj < 0.1, 'royalblue',
ifelse(resC3$log2FoldChange >= 0 & resC3$padj < 0.1, 'red2',
'black'))
keyvals.colour[is.na(keyvals.colour)] <- 'black'
names(keyvals.colour)[keyvals.colour == 'red2'] <- 'Up-regulated'
names(keyvals.colour)[keyvals.colour == 'black'] <- 'NS'
names(keyvals.colour)[keyvals.colour == 'royalblue'] <- 'Down-regulated'
Select the labels to print:
selectLab<-resC3$symbol[names(keyvals.colour) %in% c('Up-regulated', 'Down-regulated')]
selectLab<-selectLab[!is.na(selectLab)]
str(selectLab)
chr [1:16] "Msc" "Knstrn" "Ovol2" "Olfml3" "Gstm2" "Lrrc17" "Oas1b" "Lrguk" "Tspan11" "Mrgpra3" "Plat" "Tmem184c" "Ppp2r1b" ...
Plot:
EC3<-EnhancedVolcano(resC3,
subtitle = '',
lab = resC3$symbol,
x = 'log2FoldChange',
y = 'padj',
selectLab = selectLab,
title = contrast,
pCutoff = 0.0,
FCcutoff = 0.0,
pointSize = 2.0,
labSize = 4.0,
#col=c('grey30', 'grey30', 'royalblue', 'red2'),
cutoffLineType = 'blank',
cutoffLineCol = 'black',
cutoffLineWidth = 0.8,
hline = c(10e-2),
hlineCol = c('black'),
hlineType = c('longdash'),
hlineWidth = c(0.6),
vline = c(0),
vlineCol = c('black'),
vlineType = c('longdash'),
vlineWidth = c(0.6),
gridlines.major = FALSE,
gridlines.minor = FALSE,
colCustom = keyvals.colour) +
theme(axis.text.y = element_text(size=10))
Only a few are plotted.
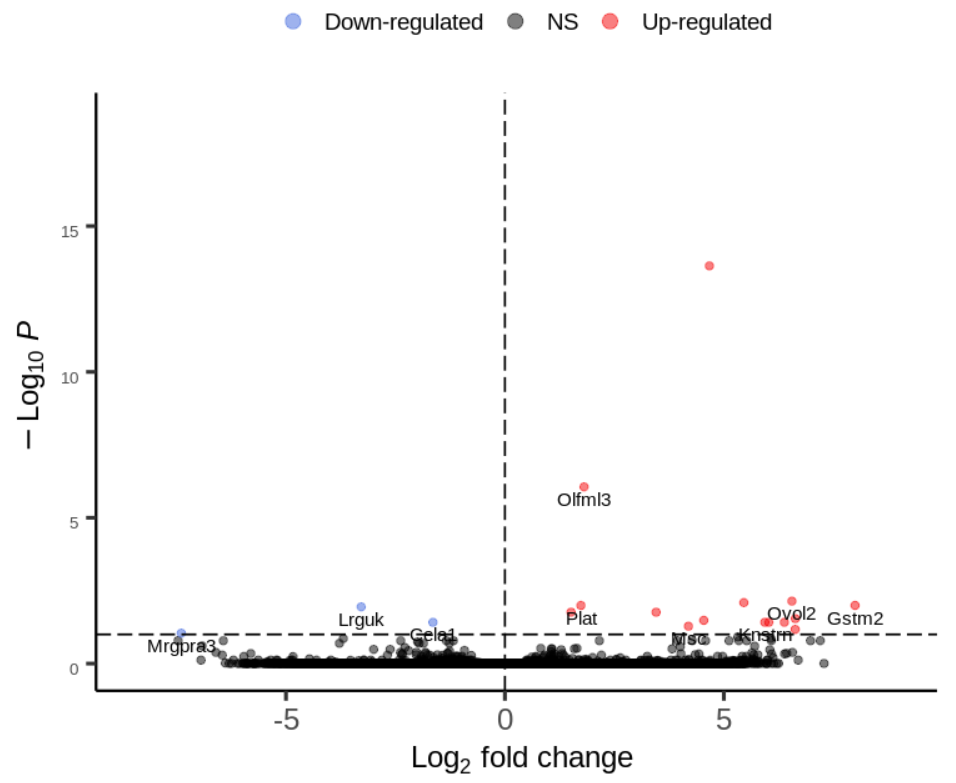
Could you help on this please? Many thanks.
sessionInfo( )
R version 4.0.0 (2020-04-24)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /g/easybuild/x86_64/CentOS/7/haswell/software/OpenBLAS/0.3.9-GCC-9.3.0/lib/libopenblas_haswellp-r0.3.9.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8 LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] ggbreak_0.0.8 biomaRt_2.44.1 DBI_1.1.0 RSQLite_2.2.0
[5] GO.db_3.11.4 mgsa_1.36.0 EnhancedVolcano_1.6.0 ggrepel_0.8.2
[9] knitr_1.28 GGally_1.5.0 gridExtra_2.3 RColorBrewer_1.1-2
[13] GenomicFeatures_1.40.1 scales_1.1.0 reshape2_1.4.4 org.Mm.eg.db_3.11.4
[17] AnnotationDbi_1.50.1 pheatmap_1.0.12 gplots_3.0.3 genefilter_1.70.0
[21] ggplot2_3.3.0 DESeq2_1.28.1 SummarizedExperiment_1.18.2 DelayedArray_0.14.0
[25] matrixStats_0.56.0 Biobase_2.48.0 GenomicRanges_1.40.0 GenomeInfoDb_1.24.2
[29] IRanges_2.22.2 S4Vectors_0.26.1 BiocGenerics_0.34.0
loaded via a namespace (and not attached):
[1] colorspace_1.4-1 ellipsis_0.3.0 XVector_0.28.0 aplot_0.1.1 rstudioapi_0.11
[6] farver_2.0.3 bit64_0.9-7 fansi_0.4.1 splines_4.0.0 cachem_1.0.6
[11] geneplotter_1.66.0 Rsamtools_2.4.0 annotate_1.66.0 dbplyr_1.4.3 compiler_4.0.0
[16] httr_1.4.2 assertthat_0.2.1 Matrix_1.3-4 fastmap_1.0.1 cli_2.0.2
[21] htmltools_0.4.0 prettyunits_1.1.1 tools_4.0.0 gtable_0.3.0 glue_1.4.0
[26] GenomeInfoDbData_1.2.3 dplyr_0.8.5 rappdirs_0.3.1 Rcpp_1.0.4.6 vctrs_0.2.4
[31] Biostrings_2.56.0 gdata_2.18.0 rtracklayer_1.48.0 xfun_0.13 stringr_1.4.0
[36] lifecycle_0.2.0 gtools_3.8.2 XML_3.99-0.3 zlibbioc_1.34.0 hms_0.5.3
[41] yaml_2.2.1 curl_4.3 memoise_2.0.0 ggfun_0.0.4 yulab.utils_0.0.4
[46] reshape_0.8.8 stringi_1.4.6 highr_0.8 caTools_1.18.0 BiocParallel_1.22.0
[51] rlang_0.4.5 pkgconfig_2.0.3 bitops_1.0-6 evaluate_0.14 lattice_0.20-41
[56] purrr_0.3.4 GenomicAlignments_1.24.0 patchwork_1.0.0 htmlwidgets_1.5.1 labeling_0.3
[61] bit_1.1-15.2 tidyselect_1.0.0 plyr_1.8.6 magrittr_1.5 R6_2.4.1
[66] pillar_1.4.3 withr_2.2.0 survival_3.1-12 RCurl_1.98-1.2 tibble_3.0.1
[71] crayon_1.3.4 KernSmooth_2.23-17 BiocFileCache_1.12.0 BisqueRNA_1.0.5 rmarkdown_2.11
[76] progress_1.2.2 locfit_1.5-9.4 grid_4.0.0 data.table_1.12.8 blob_1.2.1
[81] digest_0.6.25 xtable_1.8-4 gridGraphics_0.5-0 openssl_1.4.1 munsell_0.5.0
[86] ggplotify_0.1.0 askpass_1.1

Hi Kevin, Thanks for your reply. Here ate the requested output:
Thanks for your help.
It can be that there is no space to plot all labels. Can you try
drawConnectors = TRUEIt does the trick, thanks!