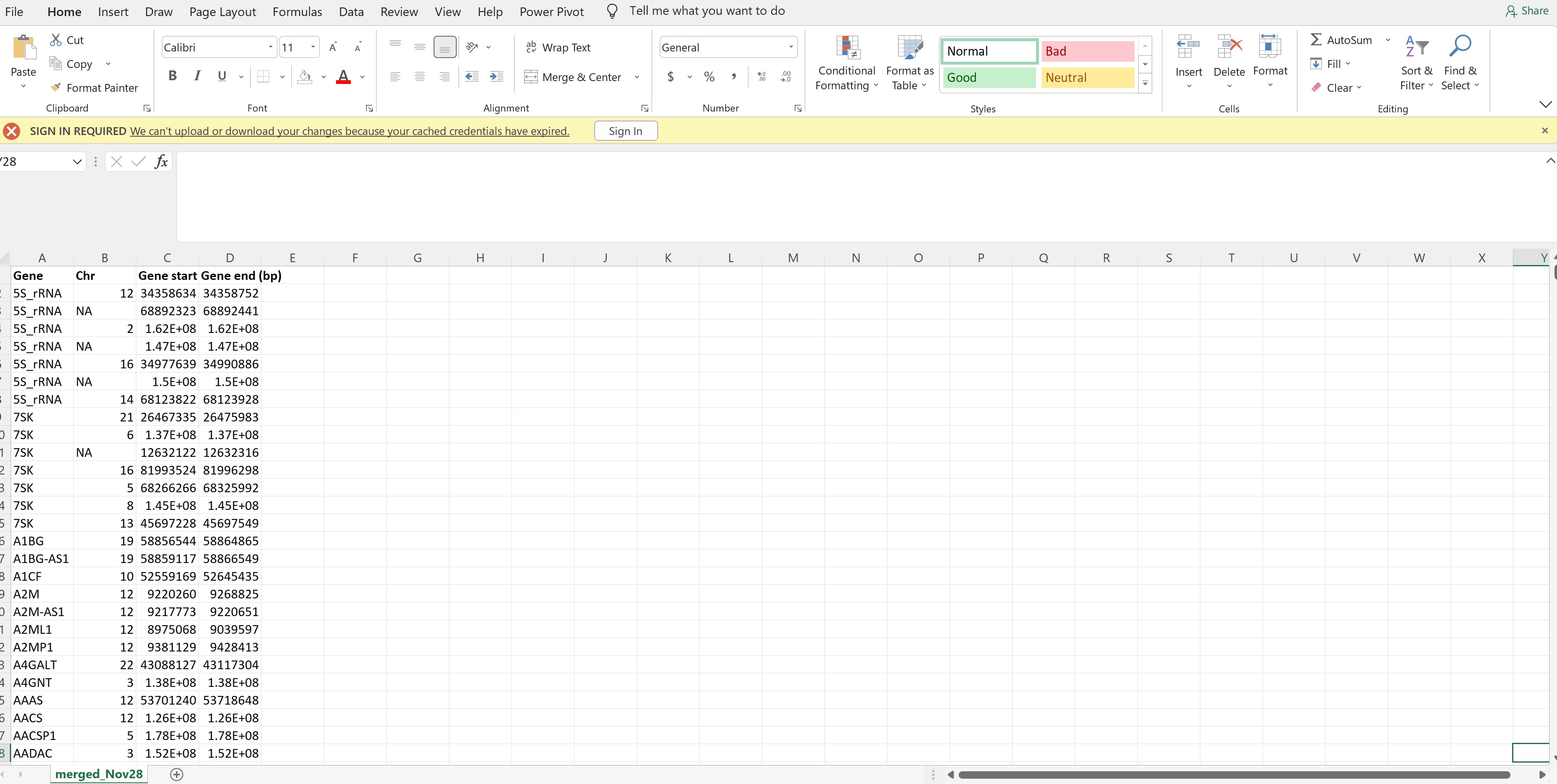
I download annotations of the chromosome start and end position from biomart , I am wondering why some of the genes appear multiple times (see picture) with different start and end positions? Also is there a way to automate deleting these gene duplicates? . \
Some genes appear multiple times because there is evidence that they arise from multiple positions in the genome. If you are asking 'where are the genes located', and we think there are multiple locations for a given gene, you will get more than one position returned. For example, there are several genes that are found on both the X and Y chromosomes (true fact!). Plus there are any number of genes that are thought to be located on various haplotypes and unplaced scaffolds.
There are functions (like duplicated or unique) that can be used to subset that list to single positions. I would also imagine there are ways to do so in Excel, since it appears that's where you have the position data currently.
If you aren't wedded to Ensembl mappings, you could always use the Homo.sapiens package, which will by default exclude any gene(s) that are found in multiple places
> library(Homo.sapiens)
> library(TxDb.Hsapiens.UCSC.hg38.knownGene)
> TxDb(Homo.sapiens) <- TxDb.Hsapiens.UCSC.hg38.knownGene
> gns <- genes(Homo.sapiens, columns = "SYMBOL")
1655 genes were dropped because they have exons located on both strands
of the same reference sequence or on more than one reference sequence,
so cannot be represented by a single genomic range.
Use 'single.strand.genes.only=FALSE' to get all the genes in a
GRangesList object, or use suppressMessages() to suppress this message.
'select()' returned 1:1 mapping between keys and columns
> gns
GRanges object with 29474 ranges and 1 metadata column:
seqnames ranges strand | SYMBOL
<Rle> <IRanges> <Rle> | <CharacterList>
1 chr19 58345178-58362751 - | A1BG
10 chr8 18386311-18401218 + | NAT2
100 chr20 44619522-44652233 - | ADA
1000 chr18 27932879-28177946 - | CDH2
100008586 chrX 49551278-49568218 + | GAGE12F
... ... ... ... . ...
9990 chr15 34229784-34338060 - | SLC12A6
9991 chr9 112217716-112333664 - | PTBP3
9992 chr21 34364006-34371381 + | KCNE2
9993 chr22 19036282-19122454 - | DGCR2
9997 chr22 50523568-50526461 - | SCO2
-------
seqinfo: 640 sequences (1 circular) from hg38 genome


OK thank James for explaining and providing this code. this is really helpful .