
Hello,
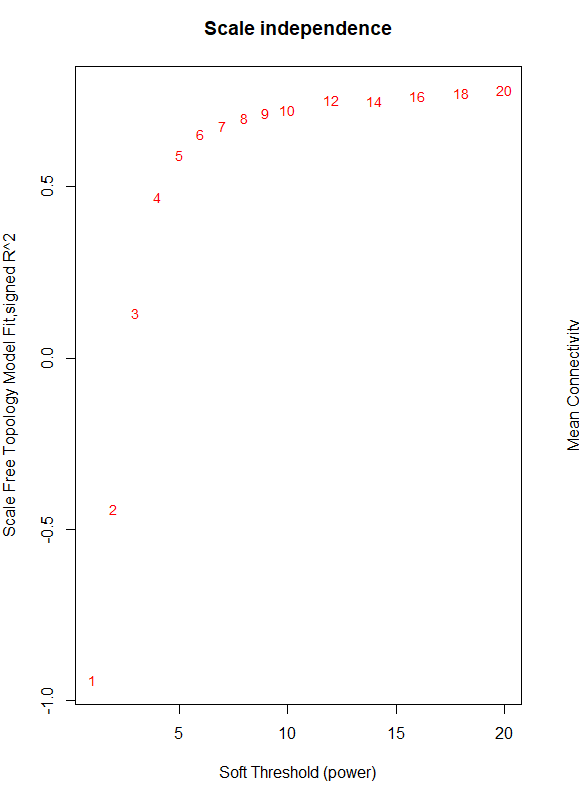
I need some guidance with finding the soft threshold for WGCNA. I have been following the tutorial and am up to the point where I design a red line that corresponds to using the R^2 cut off. I am using log (counts per million) normalized data using limma/voom for a large RNA seq experiment with many variables (including brain region, age, sex, genotype.) Limma voom is was necessary for this data set to account for the many variables. When constructing the tree diagram there are no major outliers in the data. My data does not reach the 0.9 threshold when plotting the data so the line does not fall within the graph. It seems that something is off with the power of the experiment and I am not sure how to proceed? Any help would be great! (code and graph below)
powers = c(c(1:10), seq(from = 12, to=20, by=2))
sft = pickSoftThreshold(datExpr0, powerVector = powers, verbose = 5)
sizeGrWindow(9, 5)
par(mfrow = c(1,2));
cex1 = 0.9;
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
xlab="Soft Threshold (power)",ylab="Scale Free Topology Model Fit,signed R^2",type="n",
main = paste("Scale independence"));
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels=powers,cex=cex1,col="red")
abline(h=0.90,col="red")
The output provides the following data and graph.
Power SFT.R.sq slope truncated.R.sq mean.k. median.k. max.k.
1 1 0.941 2.050 0.977 6530.0 6870.0 9170
2 2 0.442 0.415 0.735 3270.0 3440.0 5940
3 3 0.130 -0.196 0.559 1940.0 1970.0 4320
4 4 0.470 -0.535 0.736 1270.0 1220.0 3350
5 5 0.592 -0.753 0.811 884.0 795.0 2710
6 6 0.655 -0.900 0.855 644.0 539.0 2250
7 7 0.679 -1.020 0.875 486.0 377.0 1910
8 8 0.701 -1.110 0.891 376.0 270.0 1650
9 9 0.715 -1.180 0.904 297.0 196.0 1440
10 10 0.725 -1.250 0.910 239.0 145.0 1260
11 12 0.753 -1.350 0.930 162.0 83.1 1010
12 14 0.752 -1.450 0.927 114.0 50.1 820
13 16 0.763 -1.520 0.934 83.4 31.3 681
14 18 0.774 -1.570 0.943 62.5 19.8 574
15 20 0.781 -1.610 0.948 47.9 13.0 490
sessionInfo( )
```
I'm also analyzing my data using WGCNA. Not an expert. However, are you using all the genes or filter most variable genes? you can try filter most variable genes and run WGCNA again.
I am not sure if it is due to the fact that I am using pre-normalized data from my RNA-seq and not using fpkm but rather log counts per million