
Dear All,
I'm analysing my RNASeq data using WGCNA to identify genes that are co-expressed and detect modules, correlating with traits. I have done most of the analysis and identify the modules and examined the association of the modules with traits successfully. Next is to generate a summary, which contains modules with their corresponding gene ID/name and statistics.
I have normalized count data from DESeq2 with row for genename and column for sampleID. I do not have any other annotation file except the trait file, which is different dose of radiation treatment. I have attached the data frame
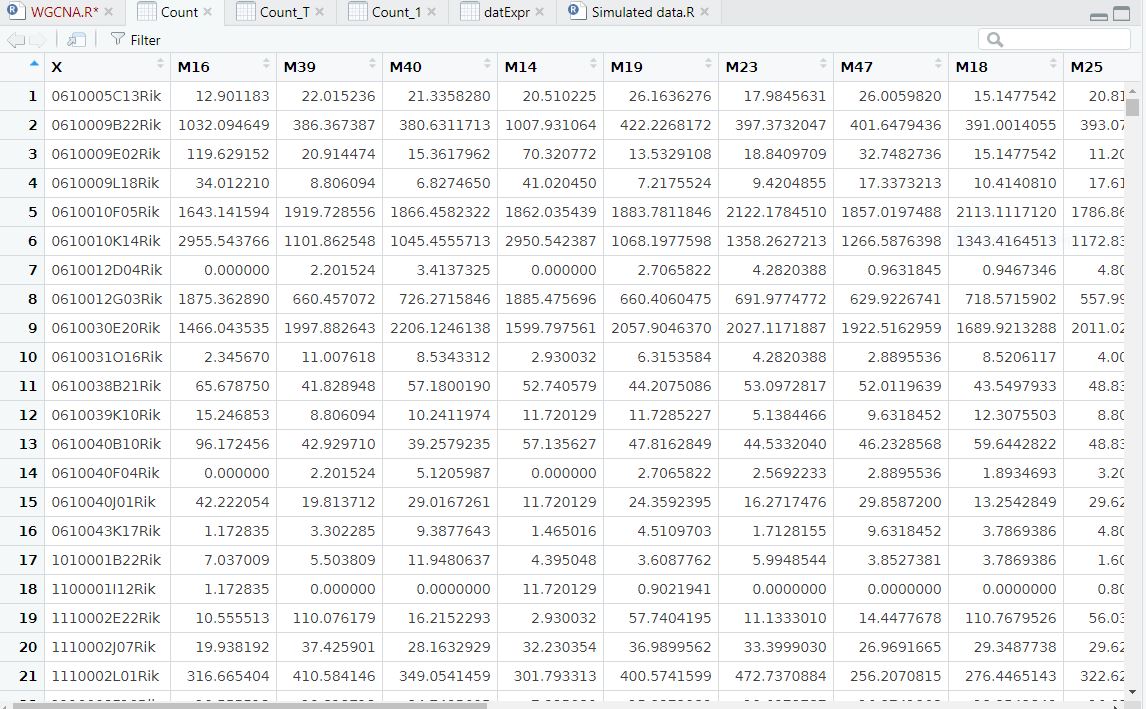
The code chunck provided in WGCNA tutorial mostly convern with microarray data and it has a comprehensive list of annotation file and I have attached the code from the tutorial in the following
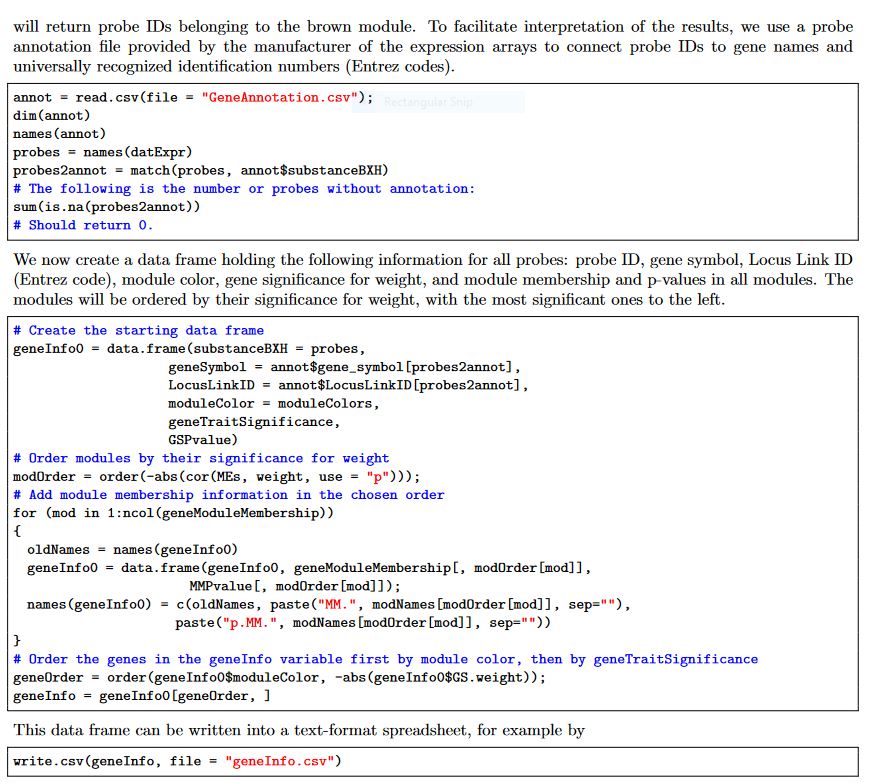
I do not have probe information, locus linkID, but only the geneID/name in the count matrix. Is there any way I could simply generate a summary with just gene name/ID in the normalized count matrix I have attached? Looking forward to hearing from the WGCNA author and others who had experience analysing WGCNA.
Kind REgards,
synat
include your problematic code here with any corresponding output
please also include the results of running the following in an R session
sessionInfo( )
```
Okay, I have successfully solved the problem. If anyone experienced the same issue, let me know.
Cheers,