
Dear author and seniors,
Hope you all are doing great. I am very new to RNASeq and DESeq2. I know that the negative Binomial (Gamma Poisson) is used to fit this RNAseq count data. All genes are assessed/fitted between two conditions and we get the basemean and log2fold change and then wald test is used to examine whether the coefficient is equal to zero if I am not wrong to determine whether the log2fold change is significant. I am so far familiar with linear rather than generalized linear model. I did a bit of poisson regression before.
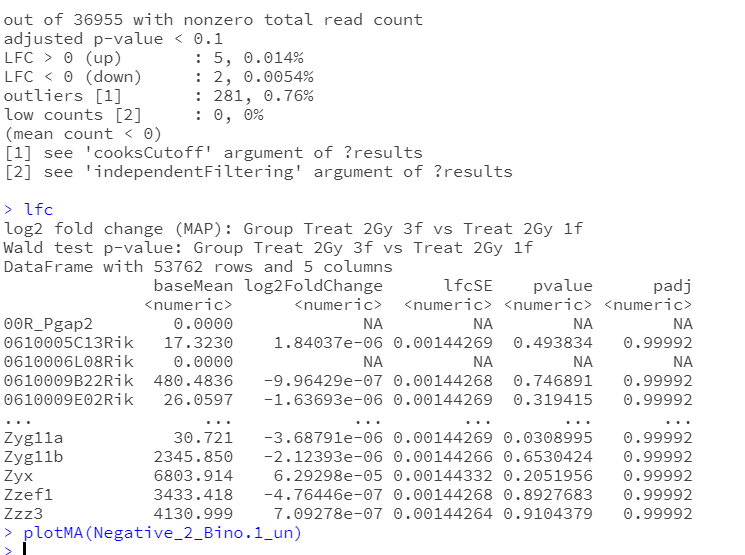
I have the attached the output of the model after fitting through DESeq2. In my output, there were five genes are upregulated and 2 genes are downregulated. Are zero is a default option of log2fold change to be considered as up and down?
Does this mean that there are seven genes in total that will have significant adjusted p-value? i recall when multiple linear regression is fitted, we get an overall p-value so we can quickly know whether at least one coefficient is not equal to zero if p-value <0.05 or vice versa or I can just skim through the output to examine how many coefficients are significant. However, with the negative 2 binomial in DESeq2, I could not find the overall p-value at all
Also, the output could not list all the genes and adjusted p-value there because there are many of them have been fitted. Therefore, I am wondering how could I know which genes have significant log2fold change by looking through the output? Hope you do not mind me with my question as I am very new to RNAseq experiment and the analysis.
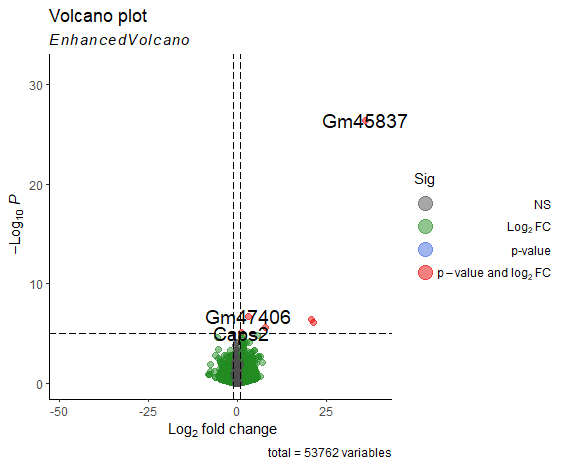
Additonally, I understand how to interpret the volcano plot. However, I am wondering whether all genes used for visialization in volcano plot? I have attached the plot, it seems not many dot points there so I am assuming only some genes are used to constructed volocano plot. Am I right? Do you think the plot looks alright. Sorry for asking and looking forward to hearing from author and seniors at your earliest convenience. Looking forward to hearing from author and seniors at your earliest convenience.
Kind Regards,
Synat
Code should be placed in three backticks as shown below
# include your problematic code here with any corresponding output
# please also include the results of running the following in an R session
sessionInfo( )

Cross posted at Biostars:
https://www.biostars.org/p/9490151/
Thank, Senior. sorry for cross posting as I was thinking in case I got different opinion, but the explanation was clear from Biostar. thank for your help.
Kind Regards,
synat