
I have been following the workflow 'RNA-seq workflow: gene-level exploratory analysis and differential expression' and I have a problem:
I tried to generate step by step the airway dataset, downloading the fastq files from https://www.ebi.ac.uk/ena/browser/view/PRJNA229998?show=reads Then I download the transcriptome from https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_29/gencode.v29.transcripts.fa.gz Then I used Salmon to perform the alignment with the same parameters as the workflow. Finally, I imported the quants files in R by 'tximeta' but the counts did not match. Moreover, when I execute the DESeq2 function, I only get 4 differential expression genes, instead of the roughly 4000 that I should get.
Is there any prefiltering process or any reason for this issue? I would like to learn these prefiltering and dataset preparation procedures to compare different bioinformatic methods with RNA-Seq.
Thanks in advance and greetings.

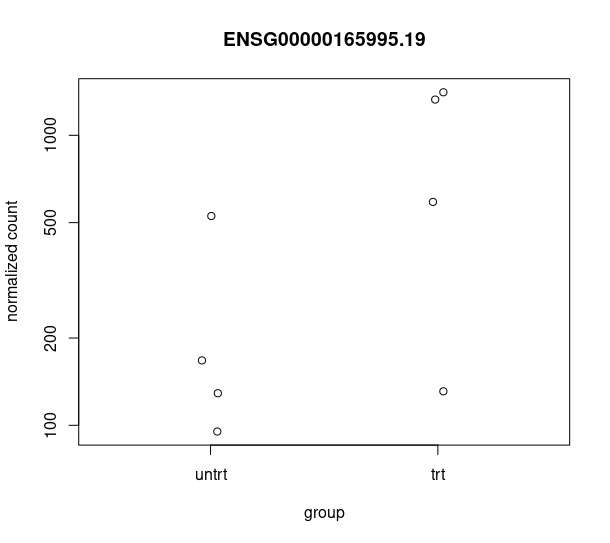
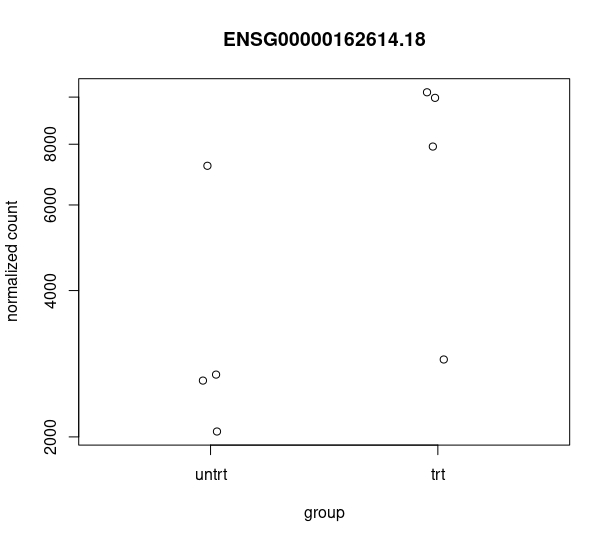
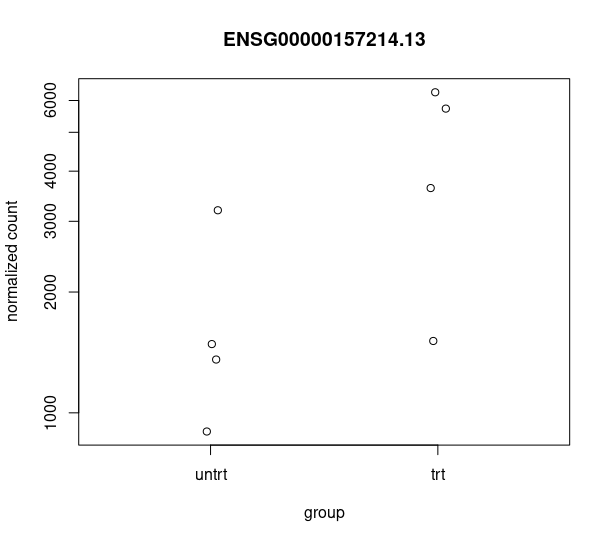
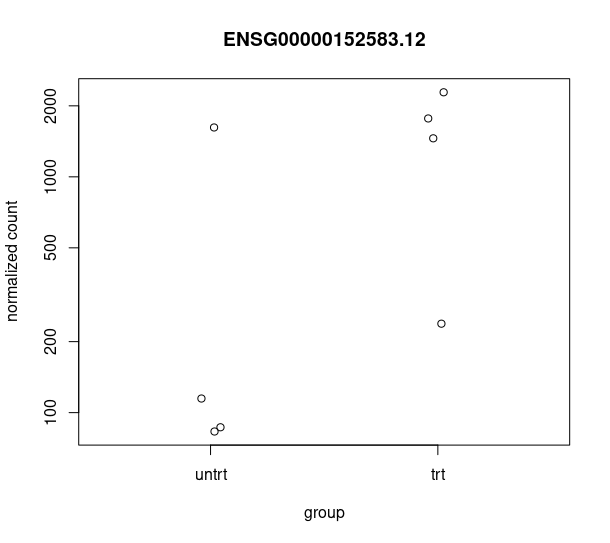
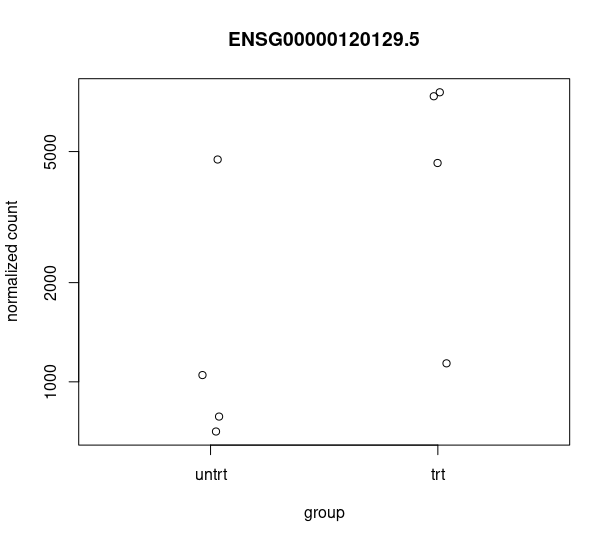

 and this csv matches with the csv from de workflow:
and this csv matches with the csv from de workflow: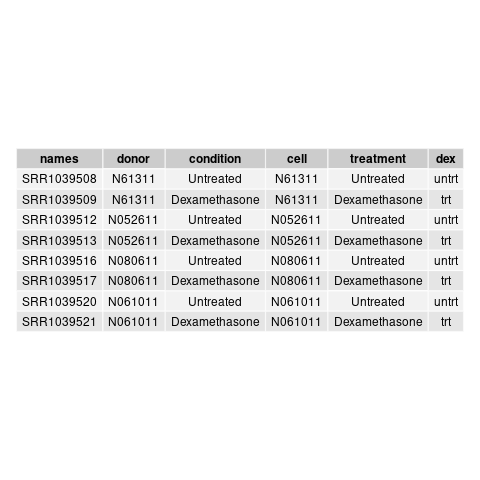
Unless you show your code, nobody can help you. Just saying what you did is no substitute for showing exactly how you did it.