Entering edit mode

I love DESeq2!
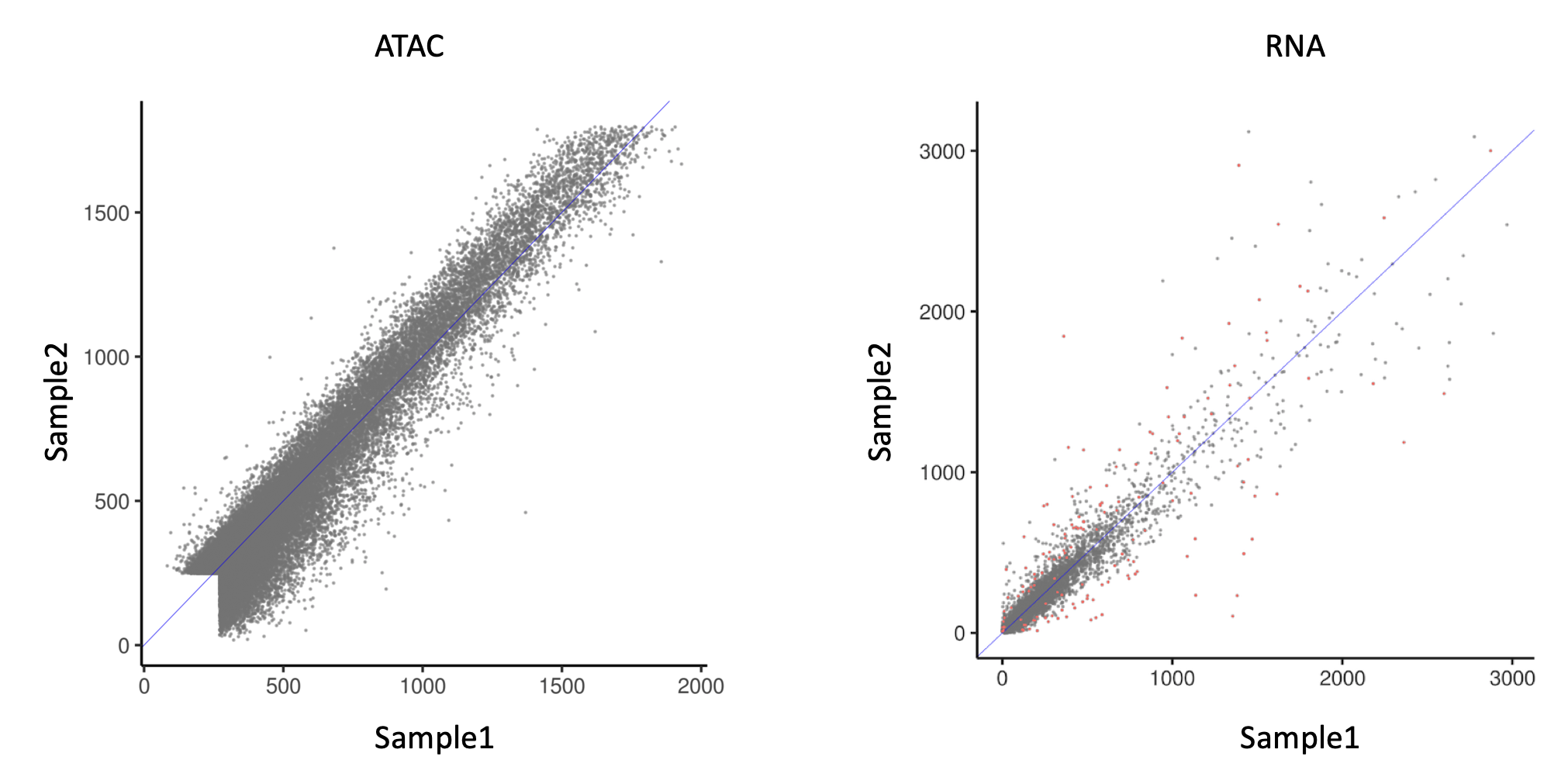
However, when using DESeq2 for ATAC analysis, I realized one potential issue. In the figure below:
Left is a scatter plot between 2 samples (biological replicates). Each point represents the read count at an ATAC peak. They are normalized by DESeq2 default normalization.
Right is a scatter plot of RNA expression between the same samples (I used multiome assay, so ATAC and RNA data are from exactly the same cells), also normalized by DESeq2 default normalization.
It's striking how these 2 assays differ. Compared to RNA, ATAC data has a striking lack of heteroskedasticity. In this case, would the statistical framework of DESeq2, which is based on a negative binomial assumption, still apply?
Thanks!

It does seem a little strange to do this, but DEseq2 does allow for a mean-dependent trend in the shared dispersion estimates, which should help compared to any tool that shrinks towards a single common dispersion estimate. Since it's not heteroskedastic, another sensible option might be limma.