
Assalam Alykom,
I'm doing a 3 factorial DESeq2 analysis with interaction terms, I want to extract results to answer specific biological questions. Here is the code for the design:
dds<-DESeqDataSetFromMatrix(countData = CountData, colData = metaData, design = ~ age + gender + viralLoad + age:viralLoad + gender:viralLoad)
dds <- DESeq(dds)
resultsNames(dds)
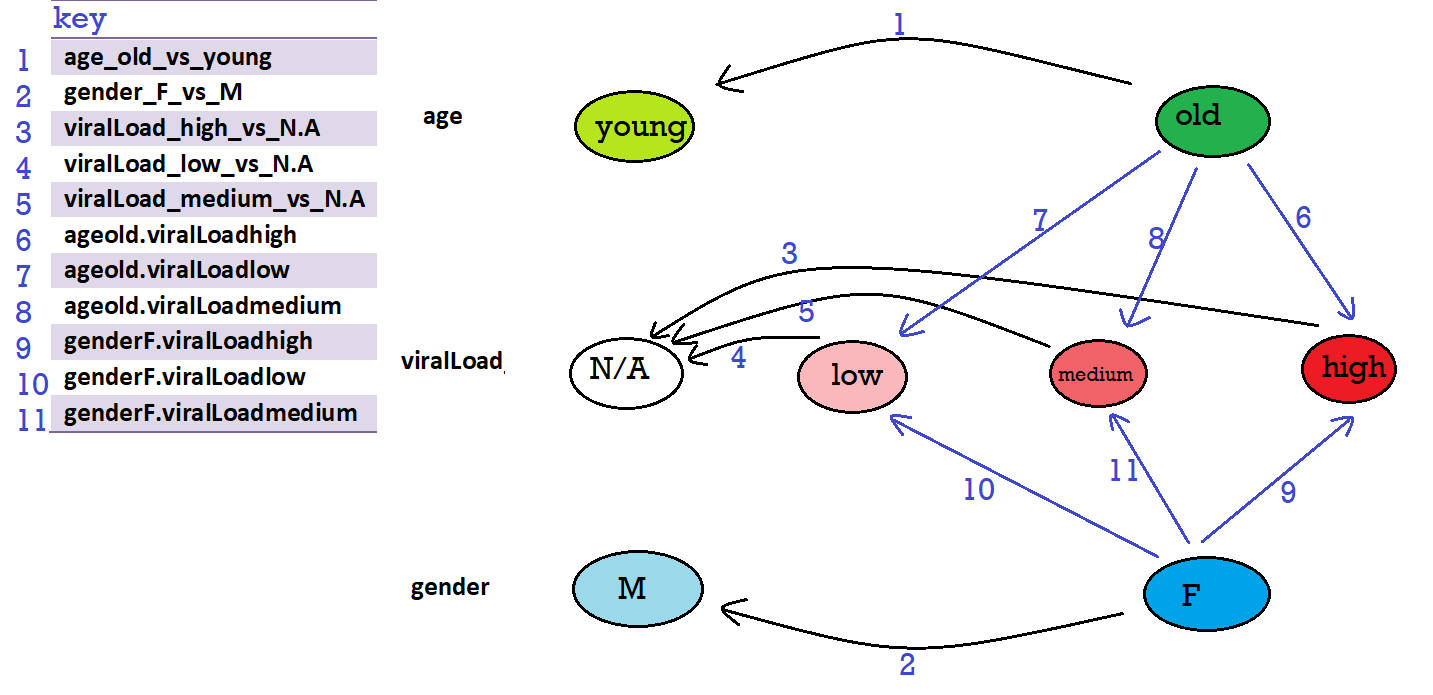
# [1] "Intercept" "age_old_vs_young" "gender_F_vs_M" "viralLoad_high_vs_N.A"
# [5] "viralLoad_low_vs_N.A" "viralLoad_medium_vs_N.A" "ageold.viralLoadhigh" "ageold.viralLoadlow"
# [9] "ageold.viralLoadmedium" "genderF.viralLoadhigh" "genderF.viralLoadlow" "genderF.viralLoadmedium"
If I want to answer the following question:
1- For females, what are the DEGs in high viral load vs. control?
Reading the example here, I drew the following design graph (reference levels are: age: young, viral load: N.A, gender: M):
res <- results(dds, contrast=c(0,0,0,1,0,0,0,0,0,1,0,0), alpha=0.05)
But what if I want to answer: 2- For males, what are the DEGs in high viral load vs. control? How should I do this? as there is no interaction term such as: genderM.viralLoadhigh in the list of resultsNames(dds). Could I use something like: viralLoad_high_vs_N.A - gender_F_vs_M - genderF.viralLoadhigh ?
sessionInfo( )
R version 4.0.4 (2021-02-15)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19042)
Matrix products: default
locale:
[1] LC_COLLATE=English_United States.1252 LC_CTYPE=English_United States.1252
[3] LC_MONETARY=English_United States.1252 LC_NUMERIC=C
[5] LC_TIME=English_United States.1252
attached base packages:
[1] stats4 parallel stats graphics grDevices utils datasets methods base
other attached packages:
[1] pheatmap_1.0.12 RColorBrewer_1.1-2 EnhancedVolcano_1.8.0
[4] ggrepel_0.9.1 forcats_0.5.1 stringr_1.4.0
[7] purrr_0.3.4 readr_2.0.1 tidyr_1.1.3
[10] tibble_3.1.3 ggplot2_3.3.5 tidyverse_1.3.1
[13] DESeq2_1.30.1 SummarizedExperiment_1.20.0 MatrixGenerics_1.2.1
[16] matrixStats_0.60.0 dplyr_1.0.7 GEOquery_2.58.0
[19] Biobase_2.50.0 rtracklayer_1.49.5 GenomicRanges_1.42.0
[22] GenomeInfoDb_1.26.7 Biostrings_2.58.0 XVector_0.30.0
[25] IRanges_2.24.1 S4Vectors_0.28.1 AnnotationHub_2.22.1
[28] BiocFileCache_1.14.0 dbplyr_2.1.1 BiocGenerics_0.36.1
loaded via a namespace (and not attached):
[1] ggbeeswarm_0.6.0 colorspace_2.0-2 ellipsis_0.3.2
[4] fs_1.5.0 rstudioapi_0.13 bit64_4.0.5
[7] interactiveDisplayBase_1.28.0 AnnotationDbi_1.52.0 fansi_0.5.0
[10] lubridate_1.7.10 xml2_1.3.2 splines_4.0.4
[13] extrafont_0.17 cachem_1.0.5 geneplotter_1.68.0
[16] knitr_1.33 jsonlite_1.7.2 Rsamtools_2.6.0
[19] Rttf2pt1_1.3.9 broom_0.7.9 annotate_1.68.0
[22] shiny_1.6.0 BiocManager_1.30.16 compiler_4.0.4
[25] httr_1.4.2 backports_1.2.1 assertthat_0.2.1
[28] Matrix_1.3-4 fastmap_1.1.0 limma_3.46.0
[31] cli_3.0.1 later_1.3.0 htmltools_0.5.1.1
[34] tools_4.0.4 gtable_0.3.0 glue_1.4.2
[37] GenomeInfoDbData_1.2.4 maps_3.3.0 rappdirs_0.3.3
[40] tinytex_0.33 Rcpp_1.0.7 cellranger_1.1.0
[43] vctrs_0.3.8 ggalt_0.4.0 extrafontdb_1.0
[46] xfun_0.23 rvest_1.0.1 mime_0.11
[49] lifecycle_1.0.0 XML_3.99-0.6 MASS_7.3-54
[52] zlibbioc_1.36.0 scales_1.1.1 vroom_1.5.4
[55] hms_1.1.0 promises_1.2.0.1 proj4_1.0-10.1
[58] yaml_2.2.1 curl_4.3.2 memoise_2.0.0
[61] ggrastr_0.2.3 stringi_1.7.4 RSQLite_2.2.7
[64] BiocVersion_3.12.0 genefilter_1.72.1 BiocParallel_1.24.1
[67] rlang_0.4.11 pkgconfig_2.0.3 bitops_1.0-7
[70] evaluate_0.14 lattice_0.20-44 GenomicAlignments_1.26.0
[73] bit_4.0.4 tidyselect_1.1.1 magrittr_2.0.1
[76] R6_2.5.1 generics_0.1.0 DelayedArray_0.16.3
[79] DBI_1.1.1 pillar_1.6.2 haven_2.4.3
[82] withr_2.4.2 ash_1.0-15 survival_3.2-11
[85] RCurl_1.98-1.3 modelr_0.1.8 crayon_1.4.1
[88] KernSmooth_2.23-20 utf8_1.2.2 tzdb_0.1.2
[91] rmarkdown_2.10 locfit_1.5-9.4 grid_4.0.4
[94] readxl_1.3.1 blob_1.2.2 reprex_2.0.1
[97] digest_0.6.27 xtable_1.8-4 httpuv_1.6.2
[100] munsell_0.5.0 beeswarm_0.4.0 vipor_0.4.5


Thanks very much