
Hi,
I am studying the ATAC-seq data analysis pipeline recently. For the pre-alignment QC step, I used FastQC and trimmomatic tools. Then more than half of the reads were reserved and mapped to the reference genome using Bowtie2. After sequence alignment, duplicated reads, mitochondrial reads, non-unique alignments and improperly mapped reads were filterd. To evaluate the quality metrics of ATAC-seq data, fragement size distribution plots were generated using ATACseqQC.
Typically, there should be a large proportion of reads with less than 100 bp, which represents the nucleosome-free region. However, in my plots, the highest peaks is not in 50~100 bp but 200 bp, which corresponds to where Tn5 inserted around a single nucleosome. And I observed that in both two biological replicates, no matter filtering or not. And I wonder Why this happend? Is there any wrong in my data processing?
Thanks in advance
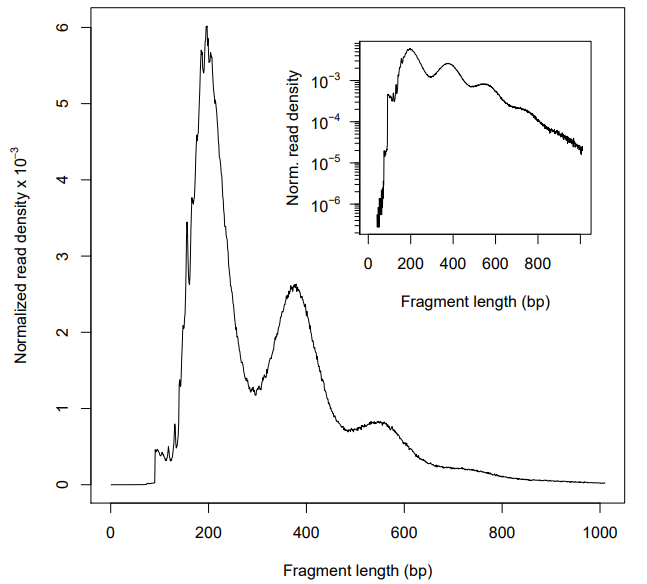
Figure 1. Fragment size distribution before filtering (There are many mitochondrial reads)
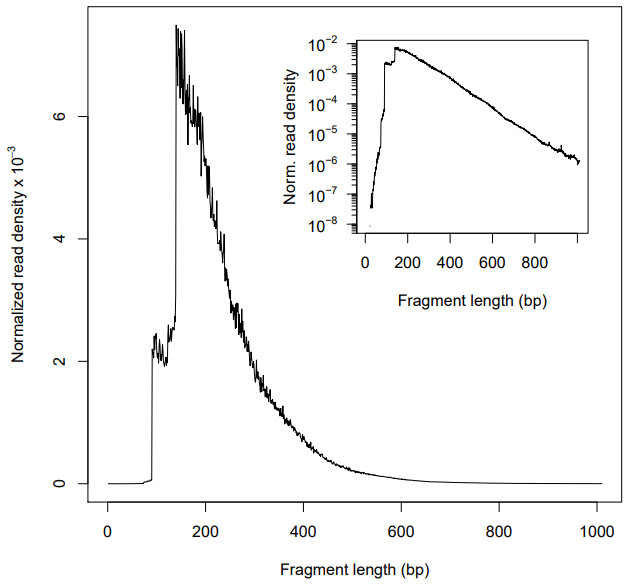
Figure 2. Fragment size distribution after filtering
# include your problematic code here with any corresponding output
# please also include the results of running the following in an R session
sessionInfo( )

What is the read size of your experiment? What kind of alignment mode were you using? End to end or local alignment? Did you tried to switch the alignment mode? Looks like the fragment less than 100bp is missing from your bam file.
Thanks for your help. It worked when I switch the end-to-end mode to local. Before filtering step, indeed, the insert fragment distribution was start from ~100 bp (Figure 3). And the quality seemed good when I only removed the mitochondrial reads (Figure 4). However, as you can see in Figure 5, the first peak of ~100 bp disappeared again when I filtered multi-mapped reads (those with MAPQ < 30, using -q in SAMtools). Does it mean that most of the short fragment (~100 bp) are non-unique alignments? If so, how to explain it? Should I remain the non-unique alignments in my bam file?
Figure 3
Figure 4
Figure 5
Try to trim the read length to 50 or 30bp before mapping and see what will happen.
Jianhong.