Entering edit mode

Guest User
★
13k
@guest-user-4897
Last seen 10.3 years ago
Hi,
I am following the protocol outlined here for analysis of single
channel Agilent microarray data:
http://matticklab.com/index.php?title=Single_channel_analysis_of_Agile
nt_microarray_data_with_Limma
I keep getting the following error message when using Limma's
read.maimages function to load my data into an RGList object:
Error in RG[[a]][, i] <- obj[, columns[[a]]] :
number of items to replace is not a multiple of replacement length
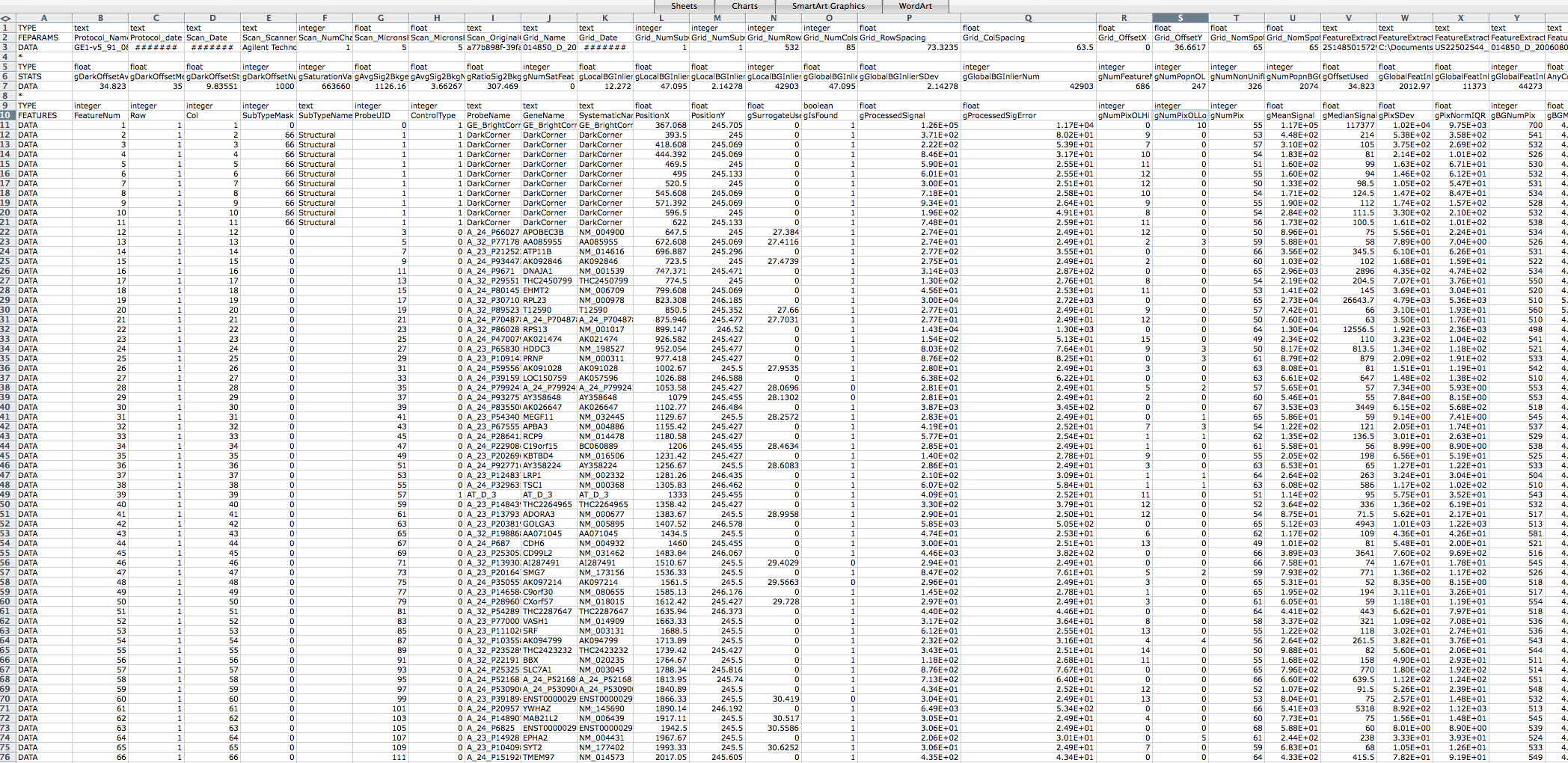
I think this may be due to my Agilent raw data txt files being in the
wrong format. I am having difficulty finding an example Agilent
feature extraction raw data txt file online to compare it to. A link
to a screen shot of one of the files I am using is below. I would
appreciate if someone could let me know if it is in the correct
format, and if not then what format it should be in to prevent the
above error message from coming up.
Thank you,
Parisa
http://www4.picturepush.com/photo/a/8322602/img/8322602.png
-- output of sessionInfo():
> sessionInfo()R version 2.13.1 (2011-07-08)
Platform: x86_64-apple-darwin9.8.0/x86_64 (64-bit)
locale:
[1] en_GB.UTF-8/en_GB.UTF-8/C/C/en_GB.UTF-8/en_GB.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] limma_3.8.3
--
Sent via the guest posting facility at bioconductor.org.


{kind=link}