
Dear List:
I want to apply quantro to RNASeq data. When I ran the quantro vignette with methylation data I got the same results as the vignette. I want to duplicate and RNASeq values and plots of an RNAseq exmaple in the quantro paper, before I apply quantro to my own data.
I applied quantro to Bottomly et al.'s dataset (PMID:21455293) downloaded from recount (PMID:22087737),
Here is the count file: Count file
Here is the phenodata file: phenodata
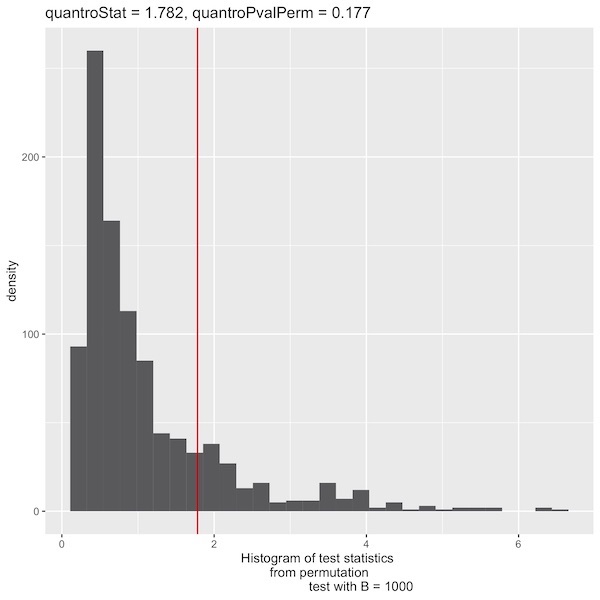
I followed the methyl protocol, with 2 extra steps (eliminating all zero genes and running the rlogTransformation) which were mentioned in the paper. I got different quantro statistics and plots than in the appendix of the paper supplement (F quantro: paper: 1.215; me: 1.78239 and P-value: paper=0.245; me=0.177 ),
Here is the density plot:
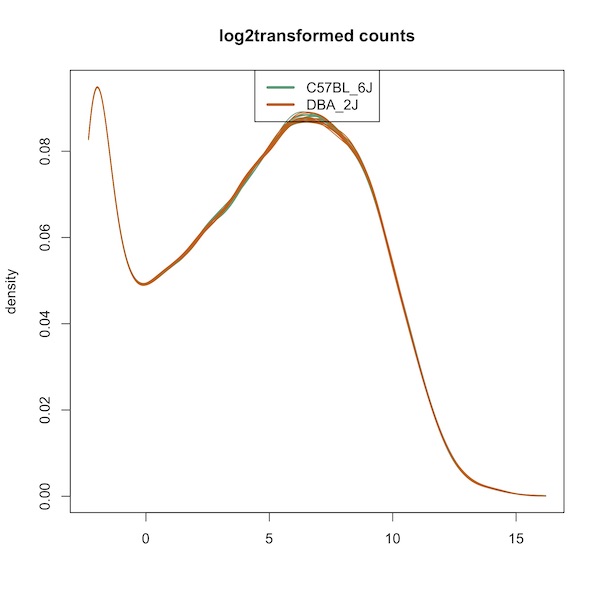
Here is the boxplot:
Here is the P-Value histogram:
Excerpts from my session:
>library(quantro)
>library(DESeq2)
>library(doParallel)
> Counts<-read.table("bottomly_count_table.txt",header=T,sep="\t",row.names=1)
> dim(Counts)
[1] 36536 21
> Counts[1:2,1:2]
SRX033480 SRX033488
ENSMUSG00000000001 369 744
ENSMUSG00000000003 0 0
> pd<-read.table("bottomly_phenodata_ed.txt",header=T,sep="\t",row.names=1)
> class(pd)
[1] "data.frame"
> dim(pd)
[1] 21 1
> pd
strain
SRX033480 C57BL_6J
SRX033488 C57BL_6J
,
.
.
SRX033494 DBA_2J
> # Make factor variable
> strain<-factor(pd$strain)
> strain[1]
[1] C57BL_6J
Levels: C57BL_6J DBA_2J
> class(strain)
[1] "factor"
> > # Make DeSeq2DataSet to input into rlogTransformation
>
> dds <- DESeqDataSetFromMatrix(countData = Counts.m,
+ colData = DataFrame(strain),
+ design = ~ strain)
> > # perform rlogTransformation, to create a DESeqTransform object
> log2transCounts<-rlogTransformation(dds,blind=FALSE)
> class(log2transCounts)
[1] "DESeqTransform"
attr(,"package")
[1] "DESeq2"
> dim(log2transCounts)
[1] 13932 21# extract log2counts matrix from DESeqTransform object
log2transCounts.m<-as.matrix(log2transCounts@assays@data@listData[[1]])
log2transCounts.m[1:2,1:2]
rownames(log2transCounts.m)<-rownames(Counts.m)
log2transCounts.m[1:2,1:2]
## ----plot-distributions-density, fig.height=5, fig.width=6-----------------
matdensity(log2transCounts.m, groupFactor = pd$strain, xlab = " ", ylab = "density", main = "log2transformed counts")
legend('top', c("C57BL_6J", "DBA_2J"), col = c(1, 2), lty = 1, lwd = 3)
> ## ----calculate-quantro----------------------------------------------------
> qtest <- quantro(object = log2transCounts.m, groupFactor = pd$strain)
[quantro] Average medians of the distributions are
equal across groups.
[quantro] Calculating the quantro test statistic.
[quantro] No permutation testing performed.
Use B > 0 for permutation testing.
> qtest
quantro: Test for global differences in distributions
nGroups: 2
nTotSamples: 21
nSamplesinGroups: 10 11
anovaPval: 0.14366
quantroStat: 1.78239
quantroPvalPerm: Use B > 0 for permutation testing.
> ## ----quantro-summary-------------------------------------------------------
> summary(qtest)
$nGroups
[1] 2
$nTotSamples
[1] 21
$nSamplesinGroups
C57BL_6J DBA_2J
10 11
>
> ## ----quantro-medians-------------------------------------------------------
> anova(qtest)
Analysis of Variance Table
Response: objectMedians
Df Sum Sq Mean Sq F value Pr(>F)
groupFactor 1 0.0005763 0.00057629 2.3265 0.1437
Residuals 19 0.0047064 0.00024771
>
> ## ----quantro-quantroStat---------------------------------------------------
> quantroStat(qtest)
[1] 1.782389
>
>
> ## ----quantro-parallel------------------------------------------------------
> registerDoParallel(cores=1)
> qtestPerm <- quantro( log2transCounts.m, groupFactor = pd$strain, B = 1000)
[quantro] Average medians of the distributions are
equal across groups.
[quantro] Calculating the quantro test statistic.
[quantro] Starting permutation testing.
[quantro] Using a single core (backend: doParallelMC,
version: 1.0.15).
> qtestPerm
quantro: Test for global differences in distributions
nGroups: 2
nTotSamples: 21
nSamplesinGroups: 10 11
anovaPval: 0.14366
quantroStat: 1.78239
quantroPvalPerm: 0.177
> ## ----quantro-plot, fig.height=5, fig.width=6-------------------------------
> quantroPlot(qtestPerm)
> sessionInfo()
R version 3.6.3 (2020-02-29)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Mojave 10.14.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets methods base
other attached packages:
[1] doParallel_1.0.15 iterators_1.0.12 foreach_1.4.7 DESeq2_1.26.0 SummarizedExperiment_1.16.1
[6] DelayedArray_0.12.3 BiocParallel_1.20.1 matrixStats_0.55.0 Biobase_2.46.0 GenomicRanges_1.38.0
[11] GenomeInfoDb_1.22.1 IRanges_2.20.2 S4Vectors_0.24.4 BiocGenerics_0.32.0 quantro_1.20.0
loaded via a namespace (and not attached):
[1] colorspace_1.4-1 siggenes_1.60.0 mclust_5.4.6 htmlTable_1.13.2 XVector_0.26.0
[6] base64enc_0.1-3 base64_2.0 rstudioapi_0.11 bit64_0.9-7 AnnotationDbi_1.48.0
[11] xml2_1.3.2 codetools_0.2-16 splines_3.6.3 scrime_1.3.5 geneplotter_1.64.0
[16] knitr_1.25 Formula_1.2-3 Rsamtools_2.2.3 annotate_1.64.0 cluster_2.1.0
[21] dbplyr_1.4.3 HDF5Array_1.14.4 readr_1.3.1 compiler_3.6.3 httr_1.4.1
[26] backports_1.1.5 assertthat_0.2.1 Matrix_1.2-18 lazyeval_0.2.2 limma_3.42.2
[31] htmltools_0.4.0 acepack_1.4.1 prettyunits_1.0.2 tools_3.6.3 gtable_0.3.0
[36] glue_1.4.0 GenomeInfoDbData_1.2.2 dplyr_0.8.5 rappdirs_0.3.1 doRNG_1.8.2
[41] Rcpp_1.0.2 bumphunter_1.28.0 vctrs_0.2.4 Biostrings_2.54.0 multtest_2.42.0
[46] preprocessCore_1.48.0 nlme_3.1-144 rtracklayer_1.46.0 DelayedMatrixStats_1.8.0 xfun_0.10
[51] stringr_1.4.0 lifecycle_0.2.0 rngtools_1.5 XML_3.98-1.20 beanplot_1.2
[56] zlibbioc_1.32.0 MASS_7.3-51.5 scales_1.0.0 hms_0.5.1 minfi_1.32.0
[61] rhdf5_2.30.1 GEOquery_2.54.1 RColorBrewer_1.1-2 curl_4.2 gridExtra_2.3
[66] memoise_1.1.0 ggplot2_3.2.1 rpart_4.1-15 biomaRt_2.42.1 reshape_0.8.8
[71] latticeExtra_0.6-28 stringi_1.4.3 RSQLite_2.1.2 genefilter_1.68.0 checkmate_1.9.4
[76] GenomicFeatures_1.38.2 rlang_0.4.5 pkgconfig_2.0.3 bitops_1.0-6 nor1mix_1.3-0
[81] lattice_0.20-38 purrr_0.3.4 Rhdf5lib_1.8.0 labeling_0.3 htmlwidgets_1.5.1
[86] GenomicAlignments_1.22.1 bit_1.1-14 tidyselect_1.0.0 plyr_1.8.4 magrittr_1.5
[91] R6_2.4.0 Hmisc_4.2-0 DBI_1.1.0 pillar_1.4.2 foreign_0.8-75
[96] survival_3.1-8 RCurl_1.95-4.12 nnet_7.3-12 tibble_2.1.3 crayon_1.3.4
[101] BiocFileCache_1.10.2 progress_1.2.2 locfit_1.5-9.1 grid_3.6.3 data.table_1.12.6
[106] blob_1.2.0 digest_0.6.25 xtable_1.8-4 tidyr_1.0.2 illuminaio_0.28.0
[111] openssl_1.4.1 munsell_0.5.0 askpass_1.1 quadprog_1.5-7
>
I would appreciate any suggestions to resolve this discrepancy.
Thanks and best wishes. Rich Richard A Friedman, PhD Columbia University

Stephanie,
I ran your script. The graphs were the same as when I did them originally. This includes the density plot, which looks very different than the one in your paper. The statistics are also different from the paper and what I did before:
F quantro: paper: 1.215; me using my script: 1.78239 me using your script: 2.0402
P-Value: paper: 0.245; me using my script: 0.177 me using your script: 0.109
The p-value discrepancies are perhaps attributable to differences in the random number seed, but the I don't think that the density plot or F quantro should be that different.
I think that something may have changed in R or the other packages you used to get this effect. If you get the time, can you please rerun the script and see what you get?
(I am entering this note under my google account name, because I cannot log in using my raf4@cumc.columbia.edu e-mail address. The login menu keeps disappearing.)
Thanks and best wishes, Rich Richard Friedman, PhD Columbia University Cancer Center
I see that you are using Bioconductor 3.10, but the paper used version 3.1 as reported in the Software section at the end of the article.
If you want to reproduce exact results, it's up to you to start with the same version numbers of software. Stephanie has provided the R script used in the paper but all the package dependencies have been updated since Bioc 3.1.
You can install Bioconductor 3.1 by downloading R 3.2 and following these legacy instructions:
http://bioconductor.org/install/#Legacy
Mike, Thank you for your note. I have been unable to get quantro working in R3.2 after considerable effort. As best I can see the stumbling block is the dependency of DESeq2 on RcppArmadillo, I could not install RcppArmadillo with BiocInstaller. I get
(I omitted the sessionInfo above to keep the post length manageable) When I try to install it from source (using the command line in MAC OS X terminal) I get
I am not clear on how to debug this error.I would appreaciate any suggestions.
That being said, I suspect that the discrepancy between my results and Stephanie's published results may lie in the DESeq2 rlogTransformation command. As applied in my initial post above, I get
Whereas the density plot in the paper is: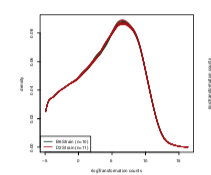
I wonder if the difference between my analysis and the published analysis lies in changes in the rlogTransformation function over time. If so, which do I believe?
Again, I regret not being able to reproduce the result in the original version of R and Bioconductor, but if you, or anyone else can consider the possibile explanation I just raised it would be helpful.
Thanks and best wishes,
Rich
Richard Friedman, PhD Columbia University
re: "I could not install RcppArmadillo with BiocInstaller"
What do you get with
install.packages("RcppArmadillo")?rlogdidn't change since its introduction really. I'd be curious to see rlog of the same data across versions, so maybe figuring out installation of RcppArmadillo is the way to go.Mike,
Thank you for your quick reply. Here is what I get:
Best wishes, Rich
The vignette for DESeq2 1.8 says it is using RcppArmadillo 0.5.600.2.0. Try grabbing that here:
https://cran.r-project.org/src/contrib/Archive/RcppArmadillo/
Mike, I did, and I got the exact same error message. I am going to reinstall the latest versions of R and Bioconductor (hopefully without overwriting 3.2.2 and try what Stephanie suggests I am willing to continue to try to get it working with 3.2.2 if you or Stephanie see any use in it. But in the meantime, I will try to reproduce the calculation that Stephanie just posted,
Thanks and best wishes, Rich
Ok, for posterity, to install a downloaded version of a package in R requires:
As this package did work with R 3.2, I don't expect you would get any error trying to install it.
Hi Rich,
I have not tried installing Bioconductor 3.1, but I did re-run the code in my GitHub repo tonight. I can confirm that I see the plots you have above (which are different than what's in the supplement) and I get the same quantro test statistic as you did above (2.0402) which is again different than what's in the paper (1.215). Unfortunately, it is unclear to me what the discrepancy is, but I agree with Michael that installing Bioconductor 3.1 would be the good thing to try.
However, just looking at the differences in the density plots, I doubt this is only related to the
rLogTransformationfunction because it looks to me like the lowly expressed genes were filtered out (as the first mode is missing). This is a complete guess, but maybe I downloaded the file locally while working with it e.g.and somehow saved a version of the dataset that had lowly expressed genes removed (?), but then changed the location back to the URL location to make the code more reproducible when I was finished with the project e.g.
To test this idea out, I tried to remove some lowly expressed genes and re-ran the code in GitHub:
The figure looks closer to what's in the supplement and the test statistic too, but not the same
Retrospectively, I recognize that I could have definitely made this code more reproducible e.g. providing a knitted HTML with
sessionInfo()describing all the details of my R session. I have learned over time to incorporate better habits for reproducibility.Stephanie,
Thank you for the clarification. I ran the script with the additional filter step in R4.0.2, Bioconductor 3.11 and got everything you got, up until the qtestPerm step. For the qtestPermstep, I got 0.176, close to what I got before. I suspect that any differences are because of the random number seed as you suggested previously. One more question: Do you recommend the kind of filtering that you just did to make the density uinimodal? It did not make a difference in the permutation p-value in this case. I ask so that I may follow the best practice.
Best wishes, Rich
Hi Rich,
If you are asking what to do with RNA-seq data as input to
quantro, yes I think it makes sense to filter out the lowly expressed genes because these genes are typically very noisy. Generally speaking, I remove lowly expressed genes for many downstream analyses, but not all. It depends on what you are wanting to do with the data. I hope this helps.All the best, Stephanie
Stephanie,
It helps. Quantro has come in handy with my dataset in showing differences in the densities across conditions. I see smooth quantile normalization in my future.
Thanks and best wishes,
Rich
Hi Rich,
I'm happy to hear this thread has been helpful, but most importantly the methods and software themselves. Wishing you all the best in your data analysis.
All the best, Stephanie