
Cross-post w/ Biostars based on commenter suggestion: https://www.biostars.org/p/345751/#345776
Hello,
I am trying to figure out how to handle biological replicates across batches in DESeq2's design matrix. They're not quite technical replicates, but trying to include the sample ID as a factor in the design matrix produces a 'Model matrix not full rank' error. What do you think I should do?
# Experiment:
We have three variables of interest in our analysis: Timepoint (A and B), Sex (M or F), and Condition (cond1 or cond2).
Note that a given sample can only have one timepoint, one sex, and one condition - so it's not like we can split the same tissue and look at it at Timepoint A and B. Each possible Timepoint x Sex x Condition has its own unique samples.
In our first sequencing batch, we collected samples for each possible combination of conditions.
In our second batch, we took some of the same RNA samples from the first sequencing batch PLUS some new RNA samples, re-generated libraries from all of these, and then sequenced.
In the end, we have a sample table that looks like this:
SAMPLE | BATCH | CONDITION | SEX | TIMEPOINT
------ ------- --------- ---- ---------
samp1 | 1 | cond1 | F | A
samp1 | 2 | cond1 | F | A
samp2 | 1 | cond1 | M | A
samp2 | 2 | cond1 | M | A
samp3 | 1 | cond1 | F | A
samp4 | 2 | cond1 | F | A
...and so forth.
# Problem
I tried to run DESeq with this:
design(dds) <- ~Genotype + Sex + Condition + SequencingBatch + Sample
and also with this:
design(dds) <- ~Genotype + Sex + Condition + SequencingBatch_Sample
...where SequencingBatch_Sample is a combination of SequencingBatch and Sample, like "samp1_1", samp1_2" for sample 1/batch1 and sample1/batch2.
These designs, of course, throw a 'Matrix not full rank' error, presumably because it is not possible to have the same sample have every possible combination of Condition/Sex/Timepoint, since each sample can only have one quality each of Condition, Sex, and Timepoint.
However, I'm not quite sure what to do instead.
Using collapseReplicates() doesn't feel appropriate because these aren't really technical replicates - a whole new library was produced for the second batch, just using the same RNA.
Doing nothing seems problematic because then the 'duplicated' signal from a subset of samples threatens to overwhelm the differential expression results.
All suggestions are very welcome - thank you!
EDIT:
Ideally, I would like to avoid collapsing Condition, Sex, and Timepoint into a single variable (e.g. Condition_Sex_Timepoint with 2 * 2 * 2 = 8 levels) because I am interested in looking at main effects and interaction effects for these variables - i.e., I'd like to understand the main effect of Sex (M vs. F) as well as the effect of Sex:Condition, and it seems harder to tease this apart if I have to do pairwise comparisons between 8 groups, e,g. Condition1+SexF+TimepointA vs. Condition1+SexF+TimepointB, Condition1+SexF+TimepointA vs. Condition1+SexM+TimepointA... etc)

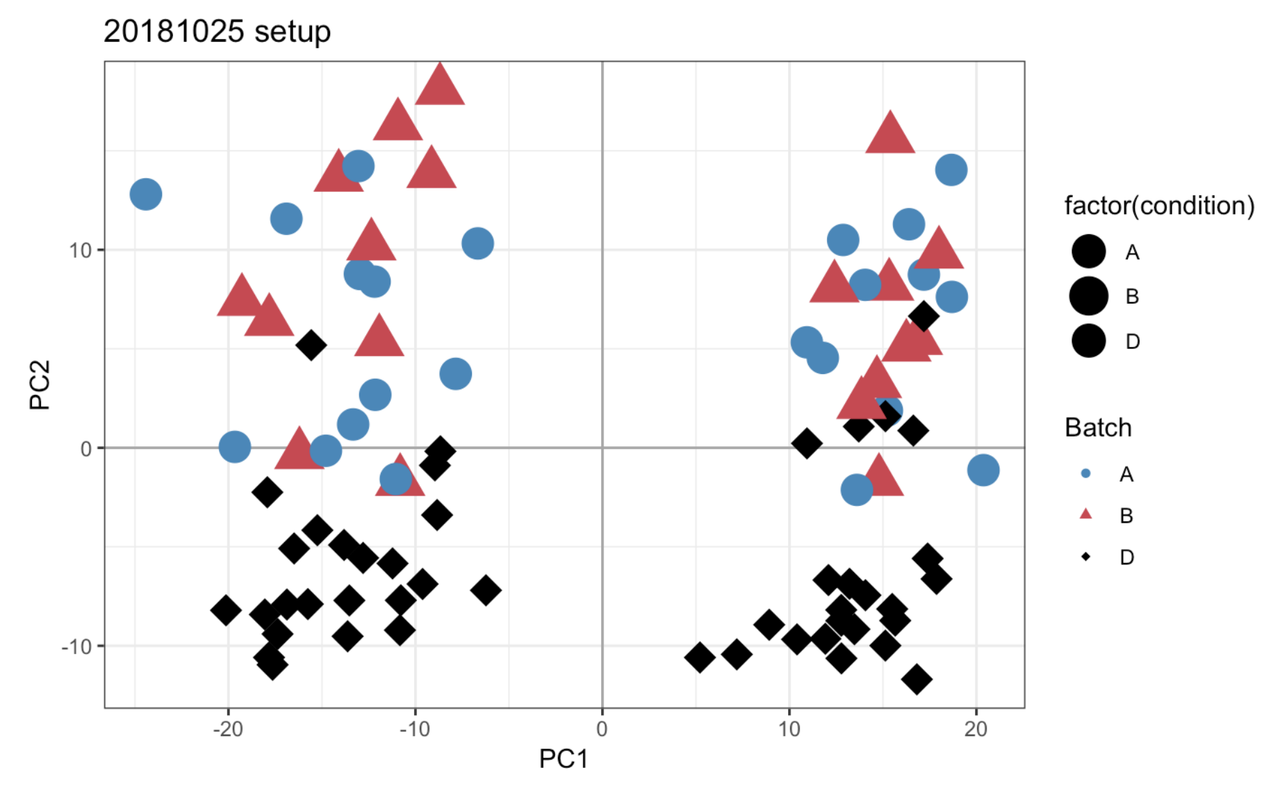
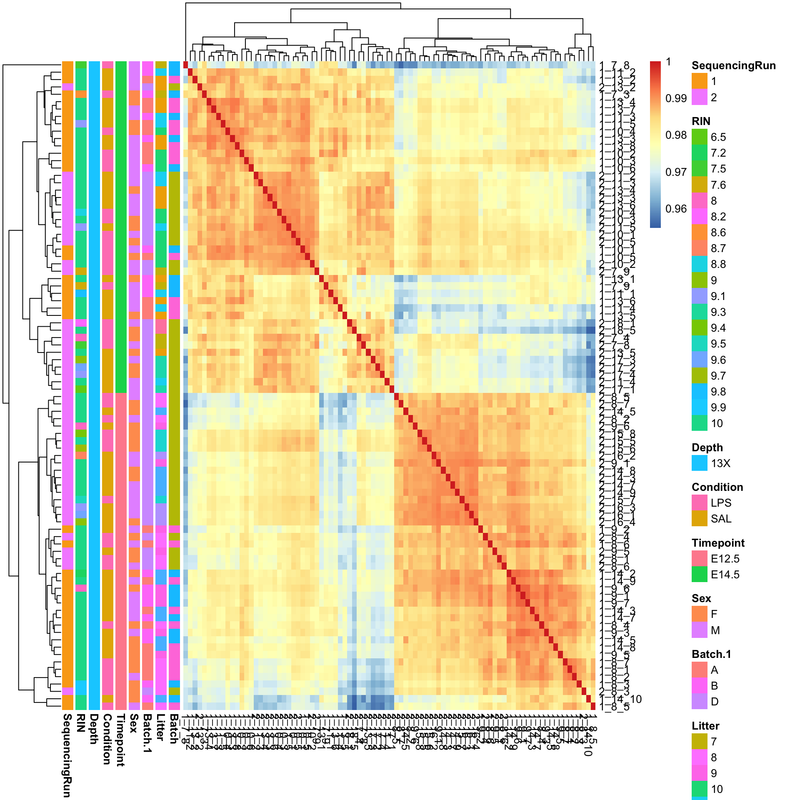
I’ll take a look, but in the meantime can you post this link on the other thread to complete the cross-link.
Ah thank you for the reminder - cross-link complete.