
Okay, I've been banging my head against this wall for too long, and it can't just be me having this problem. I'm not even sure this is the appropriate place to ask — if it's not, can someone point me to the correct forum?
I'd like to be able to use PhyloSeq's `tip_glom()` or something similar to group closely related organisms in metabarcoding studies (16S amplicons), and have annotations with particular taxa (or OTUs or RSVs... or Bioinformatically Inferred Taxon Equivalents (BITEs)) propagate to the new tip.
As an example, I've got a sample of a microbial community from before some treatment, and one from after. I'd like to annotate those taxa that persist through the treatment. Given the sampling issues etc. with NGS metabarcoding studies, I think I'd like to look at the data at a slightly higher level of relatedness: I could just use SILVA and than apply `tax_glom()` at the genus level, but it turns out more than half of my organisms get `<NA>` at the genus level. Makes more sense to tackle it phylogenetically.
Let's make an example:
```
## ================
## Toy example
## ================
## +++++
## install/load required packages:
library(phyloseq); packageVersion("phyloseq")
## install the dev build of treeio and ggtree
## (because I can't get the stable builds to play nice with each other)
devtools::install_github("GuangchuangYu/treeio")
library(treeio); packageVersion("treeio")
devtools::install_github("GuangchuangYu/ggtree")
library(ggtree); packageVersion("ggtree")
## +++++
## build an example:
## pull the data in
data("GlobalPatterns")
## prune out the first 50 taxa from the dataset
physeq = prune_taxa(taxa_names(GlobalPatterns)[1:50], GlobalPatterns)
## pull the tree out of the phyloseq object
GP.tree <- phy_tree(physeq)
## make a trait annotation for those 50 sequences
trait <- c(F,F,F,T,F,T,F,T,F,T,T,F,F,F,F,F,F,F,F,F,F,F,F,F,F,F,F,F,T,F,F,F,F,F,F,F,F,F,F,F,F,F,F,F,F,F,F,F,F,F)
trait <- as.factor(trait) # doesn't work as logical
## put the trait in a dataframe
x <- data_frame(label = GP.tree$tip.label, trait = trait)
## convert the phylo object to a treeio::treedata object
GP.tree <- treedata(phylo = GP.tree)
## add the annotation
GP.tree <- full_join(GP.tree, x, by="label")
## take a look at the tree
ggtree(GP.tree) + geom_text2(aes(subset=!isTip, label=node), hjust=-.3) + geom_tiplab(aes(color = as.numeric(trait)))
```
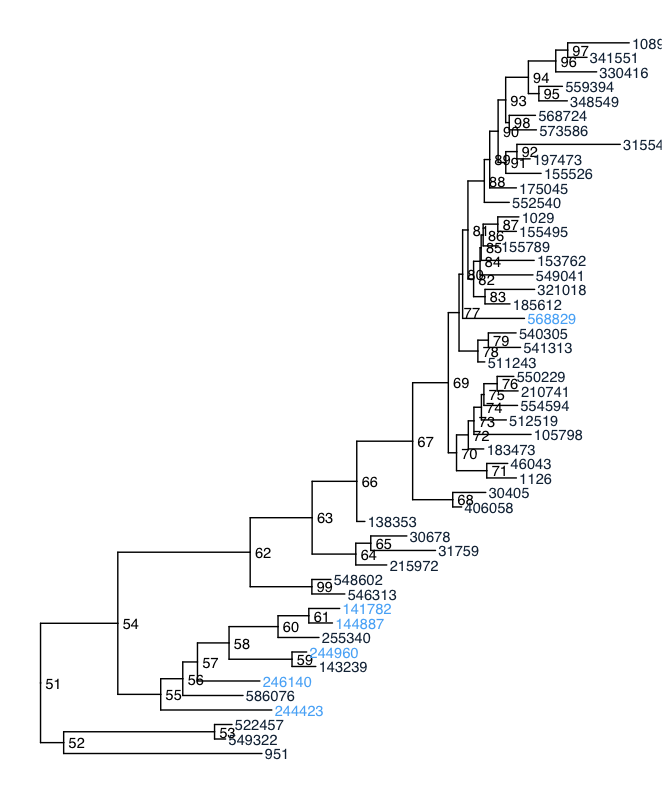
So, you can see that there's some phylogenetic signal going on here: all of the `TRUE` taxa are basically in a single clade (I did that on purpose). But, again, I'd like to simplify things by grouping related taxa — sort of like making OTUs (particularly because I'm working with _DADA2_ RSVs, and want to group them at about the species level).
_PhyloSeq_ has a great tool for this, `tip_glom()`, but I can't use the _treeio_ `treedata` object to make the `phyloseq` object; I have to convert back to an `ape::phylo` object to get back into _PhyloSeq_. And that means I lose my annotations, sadly. But, let's see what happens:
``` ## +++++ ## agglomerate tips to cluster similar taxa: ## agglomerate the tips with phyloseq GP.tree.tip <- tip_glom(as.phylo(GP.tree), h = 0.1) ## convert back to a treeio::treedata object GP.tree.tip <- treedata(phylo = GP.tree.tip) ## take a look at the tree ggtree(GP.tree.tip) + geom_text2(aes(subset=!isTip, label=node), hjust=-.3) + geom_tiplab() ```
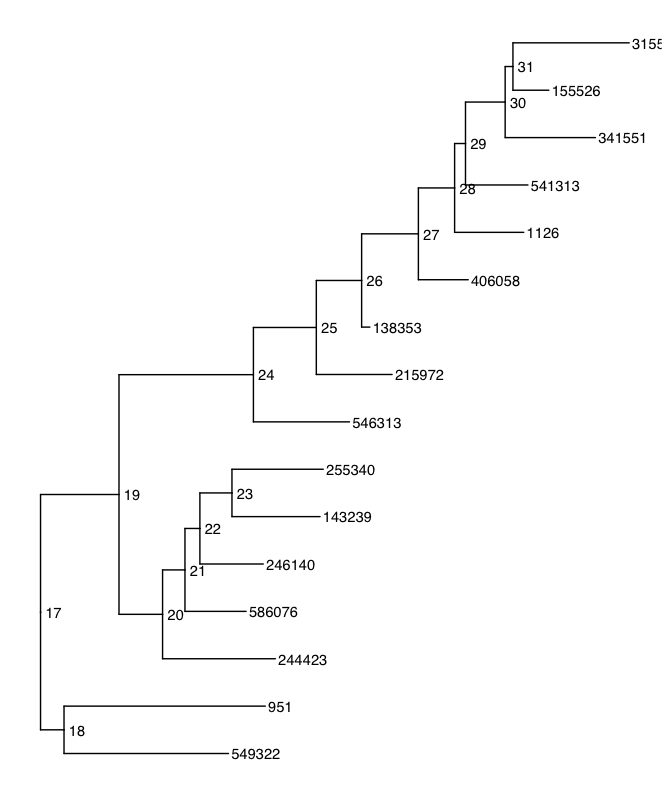
So, we end up compressing things, and it looks about like I'd like. But, the choice of which tip-labels end up as the final label for groups that are agglomerated seems a bit arbitrary — it might be the most basal taxon?
Mapping the agglomerated clades onto the initial tree, we can see that which taxa are glommed together is sometimes clear, and sometimes not. But, you can see that most of the pattern is lost!
``` ## which clades get compressed, and what ends up as the tip label? ggtree(GP.tree) + geom_text2(aes(subset=!isTip, label=node), hjust=-.3) + geom_tiplab() + geom_cladelabel(node=53, label="549322", align=TRUE, offset = 0.05) + geom_cladelabel(node=59, label="143239", align=TRUE, offset = 0.05) + geom_cladelabel(node=60, label="255340", align=TRUE, offset = 0.05) + geom_cladelabel(node=99, label="546313", align=TRUE, offset = 0.05) + geom_cladelabel(node=64, label="215972", align=TRUE, offset = 0.05) + geom_cladelabel(node=68, label="406058", align=TRUE, offset = 0.05) + geom_cladelabel(node=70, label="1126", align=TRUE, offset = 0.05) + geom_cladelabel(node=78, label="541313", align=TRUE, offset = 0.05) + #geom_cladelabel(node=59, label="group", align=TRUE, offset = 0.05) + geom_cladelabel(node=92, label="315545", align=TRUE, offset = 0.05) + geom_cladelabel(node=93, label="341551", align=TRUE, offset = 0.05) + geom_tiplab(aes(color = as.numeric(trait))) ```
So: what I want to do is really simple, and I can't believe that there's not a reasonably easy way to do it. *How can I propagate those annotations to the final tip labels?*
